Determination of linearity of measurement in MS. Linearity is the MS method's ability to provide results which are directly proportional to the concentration of the lipid analyte. Linearity depends on (a) ionization efficiency of the lipid analyte and (b) ionization behavior of lipid analyte at different concentrations depends on the used ion source. In electrospray ionization (ESI) that is used in this study, linearity holds at lower concentrations depending on (a) ion transport from ESI source to the mass analyzer and (b) the mass analyzer design and on the linearity of the detector's signal. A linearity measurement using internal standards for the molecule being analyzed has to be estimated for each experiment. As an example data set, Figure 4 shows the linearity response of PA (12:0/13:0) standard for a DIMS experiment over a concentration range spanning two orders of magnitude.
DIMS approach. Figure 5 shows all the lipid classes detected and quantified from wild type Drosophila using the global DIMS approach. Y-axis represents the mole percentage calculated on summation of all molecular species detected in each class of lipid and the percentage of total lipids detected is shown in Figure 5A. Figure 5B shows all the molecular species of PI detected and quantified and Figure 5C shows the molecular species of PC detected and quantified. PA was detected in this approach but with a smaller number of molecular species because of ion suppression. Hence, PA was detected and quantified using the DDA approach. Figure 6 shows all the molecular species of PA detected and quantified using this approach from wild type Drosophila. In all of the analyses described, the level of any specific lipid class is normalized to the total amount of lipid extracted from that sample. The total amount of lipid in a sample is estimated from the total amount of organic phosphate estimated using organic phosphate assay37.
Reverse phase liquid chromatography and tandem MS of derivatized samples. A schematic of TMS-diazomethane derivatization is shown in Figure 7A. After the derivatization of PA, ESI produces a protonated ion which upon collision induced dissociation inside the mass spectrometer methylated protonated PA molecule produces two kinds of species, which are shown in Figure 7B. First one is a neutral molecule of mass 126 Da arising from the PA headgroup and the second one is the charged diacylglycerol fragment arising from the remaining of the derivatized PA precursor ion; the mass of this fragment depends upon the constituent fatty acyl chains. Such a fragmentation pattern is true for all kinds of glycerophospholipid molecules, and one can calculate the exact mass of the derivatized precursor along with their fragment masses; such calculations for PA are shown in Table 5. Experimentally, there are two ways of setting up the MS method for detecting PA. One can detect PA by scanning for neutral losses, which is the headgroup specific neutral mass arising from the fragmentation process or one can set up multiple reaction monitoring by setting up precursors – product ion masses. In the second approach the product mass is charged diacylglycerol fragment. Figure 8 summarizes the first approach where Figure 8A shows the retention time chromatogram and Figure 8B shows the mass spectra of neutral loss scanning of 126 Da. Figure 9 shows the major PA species detected from wild type Drosophilausing the second approach.
Normal phase liquid chromatography-multiple reaction monitoring-enhanced product ion scan MS (NPLC-MRM-EPI MS). In order to establish the precise composition of the acyl chains in an individual molecule of glycerophospholipid, it is necessary to detect all the fragment ions generated from a precursor ion in a triple quadrupole mass spectrometer. To extend the process of MRM beyond precursor ion-product ion pair detection, the enhanced product ion scanning function of the machine was used. In order to obtain the precise structure of each acyl chain of an individual molecular species, i.e., regioisomers (SN1 and SN2) of PA, information dependent (IDA) auto-MS/MS experiments at different collision energy starting from -12 eV to -45 eV was performed to determine the collision energy for optimal fragmentation. IDA enables on the fly acquisition of MS/MS spectra during an MS experiment. In this experiment, MRM of known synthetic standards were used as survey scan to trigger the IDA followed by enhanced product ion (EPI) scans. As shown in Figure 10, the EPI spectrum of the m/z 549.27 [M-H]– ion of the chemically synthesized 1-dodecanoyl-2-tridecanoyl-sn-glycero-3-phosphatidic acid (12:0/13:0-PA) contains ions at m/z 199.19 ([R1COO]–) and 213.25 ([R2COO]–), corresponding to carboxylate anions arising from SN1 and SN2, respectively. Consistent with previous reports38 the intensity of the former ion is higher than the latter. The spectra also contain ions at m/z 349.21 ([M-H-R1COOH]–) and 335.16 ([M-H-R2COOH]–) corresponding to the neutral loss of the fatty acid moiety at SN1 and SN2. The intensity of the latter ion is two times more abundant than the former. The spectrum also contains ions at m/z 367.22 ([M-H-R'1CH=C=O]–) and 353.17 ([M-H-R'2CH=C=O]–) representing neutral loss of fatty acyl moiety as ketene at SN1 and at SN2, respectively. Again, the intensity of the m/z 353.17 ion is two times more than the m/z 367.22 ion. These results suggest that loss of the free fatty acid and loss of the fatty acyl ketene at SN2 is sterically more favorable than the analogous loss at SN1 and the abundance of the fragment ions m/z 335.16 ([M-H-R2COOH]–) > 353.17 ([M-H-R'2CH=C=O]–) suggest that neutral loss of the acid is a more facile process than the corresponding ketene loss. A similar pattern was observed for analogous SN1 fragments, i.e., intensity of m/z 349.21 ([M-H-R1COOH]–) > m/z 367.22 ([M-H-R'1CH=C=O]–). Therefore, the observation of the abundance of m/z 199.19 ([R1COO]–) > 213.25 ([R2COO]–) is attributed to the fact that the neutral loss of free fatty acid ([M-H-RxCOOH]–) and the neutral loss of ketene ([M-H-R'xCH=C=O]–) ions may further undergo fragmentation under the applied collision energy, CE = -39 eV, after they were formed. To validate this fragmentation pattern, a series of synthetic PA standards were characterized by EPI tandem MS to compare the types of fragment ions produced by each activation at different collision energy and evaluate the utility for determining the location of the fatty acyl chains. It was observed that for all precursor ions of PA, the fatty carboxylate anion at SN1 gives rise to the highest signal intensity at all CE other than the lowest CE of -12 eV (Figure 11). The MS/MS spectra resulting from m/z 421.24 (LPA (17:1); Figure 11A), contains fragment ions at m/z 153.02 corresponding to glycerol-3-phosphate with loss of water and m/z 267.24 corresponding to RCOO– ion. When the synthetic standard PA (17:0/14:1) was analyzed, it was observed that the highest intensity fragment ion at m/z 269.32 was accompanied by m/z 153.06, 225.28 ([R2COO]–), 361.21 ([M-H-R1COOH]–), 405.18([M-H-R2COOH]–), and 423.19([M-H-R2'CH=C=O]–) ions as shown in Figure 11B. Interestingly, the mass difference between the two highest peaks arising from fatty carboxylate ions in these two MS/MS spectra (Figure 11A,B) is of 2 Da, which corresponds to a single double bond. That means m/z 267.24 corresponding to RCOO– ion in LPA (17:1) plus 2 Da will give rise to a RCOO– ion (m/z 269.32) without the double bond, which is the case in PA (17:0/14:1). In addition to these two standards, we also analyzed the MS/MS spectra of synthetic PA (17:0/17:0; data not shown) and observed a single intense peak at m/z 269.30. To further validate this strategy for identifying the acyl chain at SN1, a similar EPI experiment was performed with a deuterated synthetic standard PA (16:0-D31/18:1), where the fatty acyl chain at SN1 alone is deuterated allowing the unambiguous identification of the peak arising from it during fragmentation. Using this standard, it was found (Figure 11C) that the two most intense peaks are at m/z 281.37 and m/z 286.36 and the latter one is at least twice more abundant than the former. Analyzing these masses clearly tells that m/z 281.37 is derived from the 18:1 carboxylate ion and since all the SN1 fatty acyl hydrogens are replaced by the heavier deuterium isotope, there is an extra mass of 31 added to the m/z of 16:0 carboxylate ion (255.36 + 31) giving rise to the peak at m/z 286.36 (Figure 11C). Thus, the potential fragment masses arising from IDA based EPI scans could be used in addition to the MRM to identify and quantify individual molecular species of PA and CE = -39 eV can be used as the optimal fragmentation energy.
We performed similar experiments with Drosophila head lipid extracts. A representative molecule of precursor mass 699.24, which is a PA (36:2), is shown in Figure 11D. As shown in Figure 11D, the tandem spectrum of the m/z 699.32 ion in EPI scan contains ions at m/z 281.30 (18:1 carboxylate anion), 283.35 (18:0 carboxylate anion), and 279.30 (18:2 carboxylate anion). The relative intensity profile of fatty acid fragments (carboxylate anion) as observed from the spectra is as follows: I281.30 > I283.35 > I279.30. From this intensity profile, it was concluded that there are two individual acyl chain specific molecules within PA (36:2) and the exact position of these fatty acyl components will be 18:1 in SN1 with 18:0 in SN2 (I281.30 > I283.35) and 18:0 in SN1 with 18:2 in SN2 (I283.35 > I279.30).
The individual extracted ion chromatogram (XIC) of all the PA molecules is shown in Figure 12. To set up these MRM-based transitions in experimental setup, a user-friendly database can be found at 31. Using this approach, we analyzed head extracts from wild type and a loss-of-function mutant of Drosophila phospholipase D (dPld3.1)23. The results are summarized in Figure 13. Using direct infusion MS, we have previously identified and confirmed the existence of 15 molecules of PA from Drosophila head extracts and seven from retinal extract23. One can quantify the level of each PA species by comparing the monoisotopic MS1 intensity of the corresponding peaks to that of the internal standard. While this represents a substantial increase over the number of PA species previously reported from Drosophila tissues15, this type of analysis is limited in its ability to precisely identify the composition of each acyl chain of an individual molecular species, i.e., regioisomers (SN1 and SN2). Using the method described here, we revisited the analysis of the levels and composition of PA in head extracts from wild type and a loss-of-function mutant of Drosophila phospholipase D (dPld3.1). Consistent with our previous analysis using high resolution mass spectrometry (HRMS)23, we found that the total amount of PA is significantly lower in dPld3.1 compared to wildtype as shown in Figure 13D. Further, using this new method, we can accurately identify individual species of PA, establishing the unique acyl chain composition (the total number of carbon atoms and double bonds in each fatty acyl chain) at SN1 and SN2 (Figure 13B). We were able to establish that in Drosophila heads, PA is a complex mixture of 36 individual molecular species with unique acyl chain compositions (Figure 13B). Palmitic (16:0), palmitoleic (16:1), oleic (18:1), linoleic (18:2), and linolenic (18:3) fatty acyl chains are found to be populated in PA from Drosophila heads. The most abundant PA species are PA (16:1/18:1), PA (16:0/18:2), PA (18:2/20:0), PA (18:1/18:1), PA (18:2/18:2), PA (18:2/18:3), PA (18:1/18:2), and PA (18:3/20:0; Figure 13B). We also determined the distributions of combined fatty acid chain length (Figure 13A) and total number of double bonds in the two fatty acyl chains (Figure 13C) and found that the C36 series is the most populated as shown in Figure 13A and the combined fatty acid chain in SN1 and SN2 containing two double bonds are most populated as shown in Figure 13C.
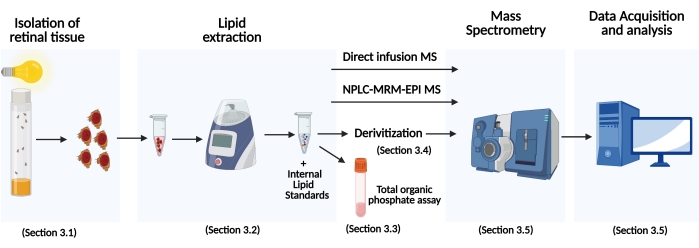
Figure 1: Schematic showing a typical workflow of MS based lipid analysis of Drosophila tissue samples. The section number in the article where each step is described is shown in the figure. Abbreviations: MS = Mass spectrometry; NPLC-MRM-EPI-MS = normal phase liquid chromatography-multiple reaction monitoring-enhanced product ion scan-MS. Please click here to view a larger version of this figure.
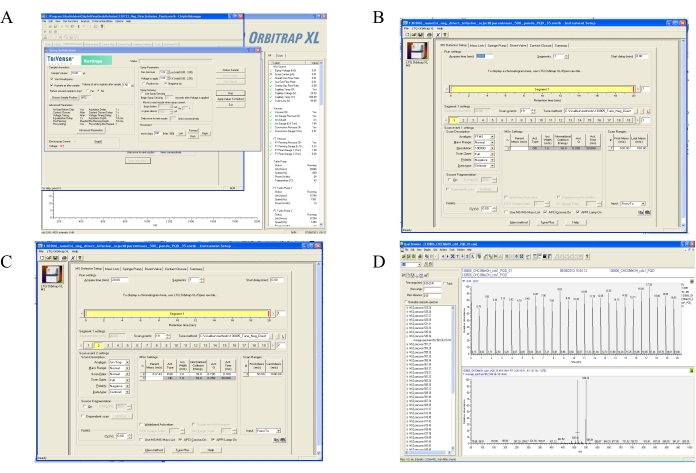
Figure 2: Mass spectrometry instrument set up for DIMS method. (A) An example of the software interface showing set up of ESI parameters on the MS used in the DIMS method. The ESI parameters such as gas pressure, voltage, and the polarity are shown in the screenshot. (B) A screenshot of the software interface of DIMS – DDA set-up on the MS software for high-resolution MS is shown. This screenshot shows the window for MS1, which is a high resolution of the Fourier transform mass spectrometry (FTMS) mode. Parameters such as run time, FTMS resolution, mass range, and polarity are set up using this interface. (C) A screenshot of the software interface of DIMS – DDA set-up on the mass spectrometer for PA analysis is shown. In this section, the analyzer should be selected as ion trap. In a step-wise manner the precursor masses of different MS1 based PA molecular ions should be given. The collision induced dissociation (CID) parameters should be given. CID parameters is given for the dissociation of lipid analyte inside mass spectrometer. Mass range should start from 50 to capture all the fragment ions. (D) An example of initial data acquired in a DIMS-DDA based experimental outcome is shown on the software interface of the mass spectrometer. The upper panel shows the chromatogram; x-axis is the elution time and y-axis is the relative abundance of the ion signal. The x-axis shows the m/z and the y-axis shows the relative abundance of an individual PA molecule. Please click here to view a larger version of this figure.
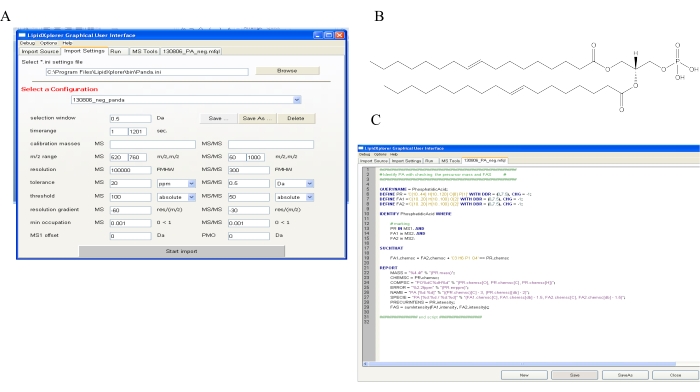
Figure 3: Data analysis of data obtained from the DIMS method. (A) A Lipidxplorer software analysis window depicting the data import settings is shown. (B) A representative chemical structure of PA (16:1/18:1) is shown. This chemical structure helps in formulating molecular fragmentation query language (MFQL) for the detection of PA from experimental runs. (C) An example MFQL for a PA molecule is shown. Please click here to view a larger version of this figure.
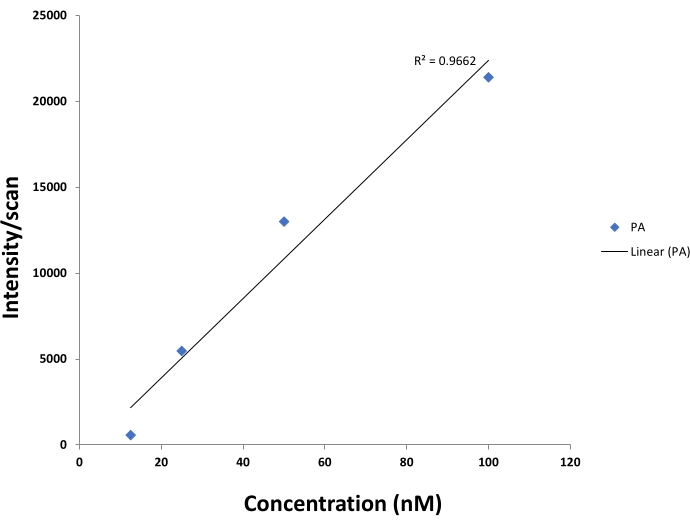
Figure 4: A linear dose-response curve observed for PA (12:0/13:0) by the DIMS approach. The y-axis represents the observed intensity per scan and the x-axis represents the concentration in nM. Effect of concentration of PA (12:0/13:0) on instrument response. A 200 nM concentration of PA (12:0/13:0) was prepared and diluted to different concentrations, and then analyzed in direct infusion mass spectrometry. Several scans were obtained. Y-axis in the plot is showing the intensity per scan. The data points were fitted linearly and a R2 of 0.9662 was obtained. Please click here to view a larger version of this figure.
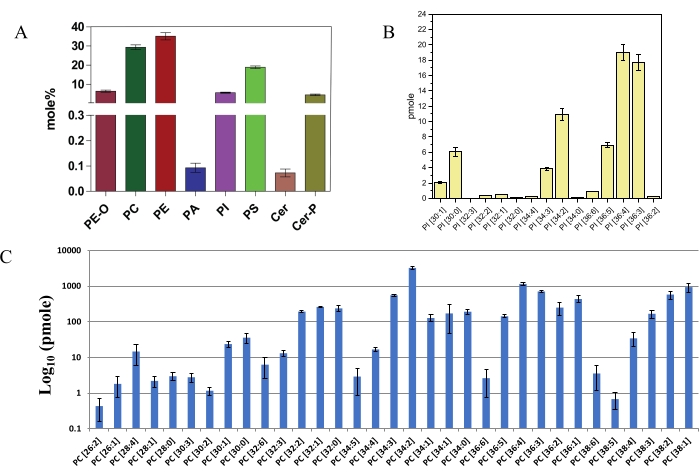
Figure 5: Major lipid classes identified and quantified using DIMS. (A) Major lipid classes measured using DIMS from wild type Drosophila head lipid extracts. X-axis represents individual lipid classes. Total lipid class abundance is presented as mol% of total lipids in y-axis. Error bars indicate the mean ± standard error of mean (SEM) from three separate analyses (n = 3). (B) PI molecules identified using DIMS and quantified using internal standard from 10 wild type Drosophila heads. X-axis represents individual PI molecular species detected. Y-axis represents the amount of each individual PI species in pmole. Error bars indicate the mean ± SEM from three separate analyses. (C) PC molecules identified using DIMS and quantified using internal standard in wild type Drosophila head extract. X-axis represents individual molecular species of PC. Y-axis represents the amount of each individual PC species from 10 heads in pmole in log10 scale. Error bars represent mean ± SEM from three separate analyses. Please click here to view a larger version of this figure.
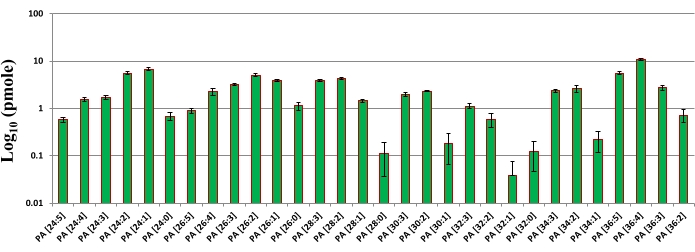
Figure 6: PA molecules identified using DIMS-DDA method and quantified using internal standard in wild type Drosophila head extract. X-axis represents different PA molecular species. Y-axis represents the amount of each individual PA molecules in pmole in log10 scale. Error bars indicate the mean ± SEM from three separate analyses (n = 3). Each individual molecular species of PA was quantified based on the intensity and amount of internal standard PA (12:0/13:0). In this approach, the exact composition of fatty acyl chain cannot be determined and hence the molecular species of PA is represented as (total number of carbon in two fatty acyl chain:total number of double bonds in two fatty acyl chain). As can be seen from the plot, PA (36:4) is the most abundant and PA (32:1) is the least abundant. Please click here to view a larger version of this figure.
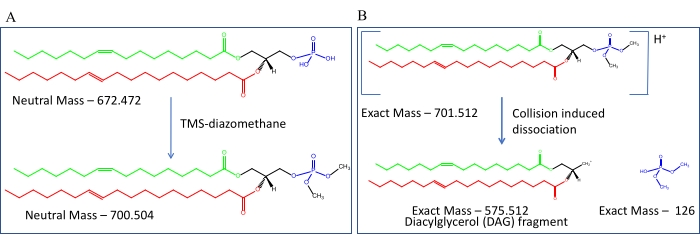
Figure 7: Chemical modification of glycerophospholipids using trimethylsilyl diazomethane (TMSD). (A) A representative PA species (16:1/18:1) with acyl chains in red and green color. After derivatization with TMSD, PA gets methylation of the hydroxyl groups attached to the phosphate groups occurs. The mass of PA before and after derivatization is shown. (B) The fragmentation of the methylated parent mass generates a neutral methylated head group and a charged diacylglycerol fragment. The mass of the precursor molecule and the fragments induced by CID is shown. The mass of the neutral methylated headgroup can be used for neutral loss scanning MS. The charged fragment mass can be used to quantify the red and green acyl chain-containing PA species by the MRM method described in the main text. Please click here to view a larger version of this figure.

Figure 8: Detection of different PA molecular species by scanning for neutral loss of 126 Da in UPLC-MS of derivatized sample. (A) Retention time chromatogram of neutral loss of 126 Da. X-axis shows retention time (min) and y-axis shows the intensity of the ion current. The box shows the region in retention time chromatogram where PA has been detected in the mass spectrometer. (B) Mass spectra of neutral loss scanning of 126 Da. The detected masses of DAG fragment after neutral loss of 126 Da of individual PA molecules are shown on top of each peak. X-axis show mass (m/z). Y-axis depicts the intensity of the ion current in counts per second unit. Each number in this plot correspond to different molecular species of PA molecules. To emphasize this, three representative arrows indicating individual PA species are shown in the figure. Please click here to view a larger version of this figure.

Figure 9: Major PA molecules identified using post TMSD derivatisation RPLC-MRM and quantified using internal standard in wild type Drosophila head extract. X-axis represents different PA molecular species. Y-axis represents the amount of each individual PA molecules in pmole per nmole total lipid phosphate. The values are mean of the samples with the error bars indicating ± SEM from three separate analyses (n = 3). Each individual molecular species of PA was quantified based on the intensity and amount of internal standard PA (17:0/14:1). In this approach, the exact composition of fatty acyl chain cannot be determined and hence the molecular species of PA is represented as (total number of carbon in two fatty acyl chain:total number of double bonds in two fatty acyl chain). As can be seen from the plot, PA (36:4), PA (36:5), PA (38:5) are the most abundant and PA containing the total number of carbon in two fatty acyl chain less than 30 are less abundant. Please click here to view a larger version of this figure.
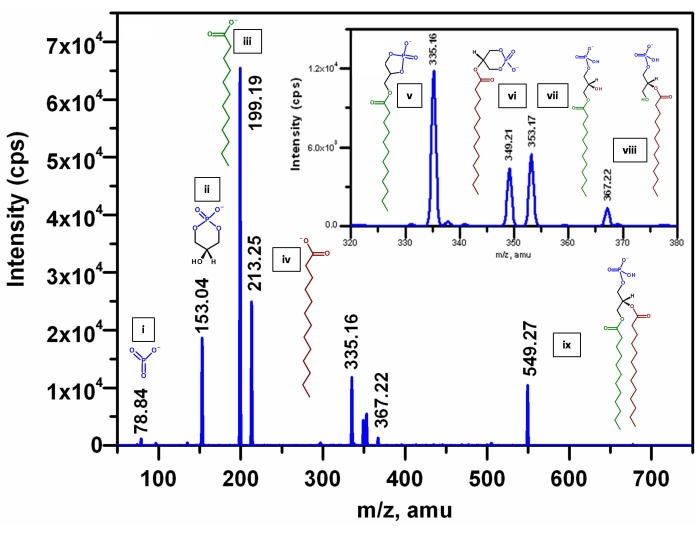
Figure 10: Enhanced product ion (EPI) spectra of [M-H]– ion of synthetic PA (12:0/13:0) standard. The fragments are listed here: (i) PO3– ion (from phosphate) at m/z 78.84 Th, (ii) glycerol-3-phosphate ion with loss of H2O at m/z 153.04 Th, (iii) SN1 RCOO– ion at m/z 199.19 Th, (iv) SN2 RCOO– ion at m/z 213.25 Th, (v) neutral loss of SN2 RCOOH group from [M-H]– at m/z 335.16 Th, (vi) neutral loss of SN1 RCOOH group from [M-H]– at m/z 349.21 Th, (vii) loss of SN2 acyl chain as ketene (RCH=C=O) from [M-H]– at m/z 353.17 Th, (viii) loss of SN1 acyl chain as ketene (RCH=C=O) from [M-H]– at m/z 367.22 Th, (ix) Precursor ion [M-H]– at m/z 549.27 The x-axis shows the m/z values and the y-axis shows intensity of the ion current in counts per second (cps). Please click here to view a larger version of this figure.
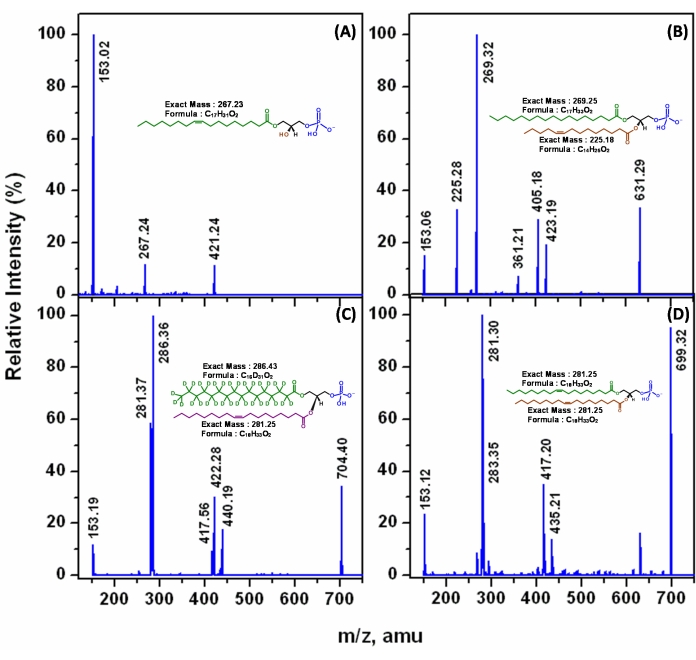
Figure 11: Enhanced product ion (EPI) spectra. EPI spectra of (A) precursor of 421.24 Th of synthetic LPA (17:1), (B) precursor of 631.29 Th of synthetic PA (17:0/14:1), (C) precursor of 704.40 Th of synthetic PA (16:0-D31/18:1), (D) precursor of 699.32 Th of tissue sample PA (18:1/18:1). Fatty acid carboxylate ions observed are as follows in (A) m/z 267.24 (SN1), (B) m/z 269.33 (SN1) and 225.28 (SN2), (C) m/z 286.36 (SN1) and 281.37 (SN2), (D) m/z 281.30 (SN1 and SN2) and 283.35 (SN1) and 279.30 (SN2). The prediction of SN1 fatty acid carboxylate ions is based on higher intensity, compared to their SN2 counterpart. Please click here to view a larger version of this figure.

Figure 12: Detection of PA by NPLC-MRM-EPI method. Extracted ion chromatograms (XICs) of detected PA molecules using specific MRM set up for precursor mass and their corresponding SN1 fragment mass are shown in the plot. The x-axis represents time in min and y-axis represents ion current, i.e., intensity in cps (counts per second). Please click here to view a larger version of this figure.
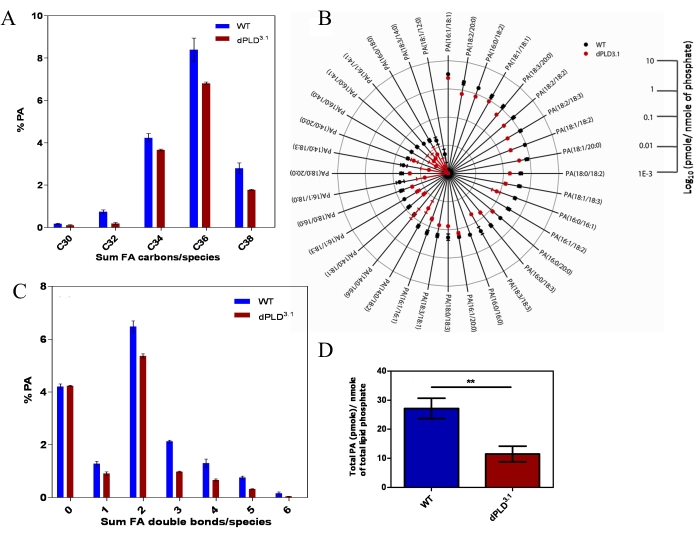
Figure 13: Results from NPLC-MRM-EPI method for PA. (A) The distributions of combined fatty acid (SN1 + SN2) chain length. The abundance of each is presented as a percent relative to total PA in its category. (B) Radar chart of individual molecular species of PA quantified using NPLC-MRM-EPI. The value of each PA species (pmole/nmole of total lipid phosphate) is depicted by the node (anchor) on the spoke (axis). A line is then drawn which connects the data values for each spoke. All data are plotted in log10 scale. N = 9. (C) The distribution of combined unsaturation (SN1 + SN2) in PA. The abundance of each is presented as a percentage relative to the total PA in its category. (D) Total phosphatidic acid levels in head extract of wildtype control and dPLD3.1 obtained from NPLC-MRM-EPI experiments. The x-axis represents the genotypes, and the y-axis shows the level of PA normalized to total lipid phosphate content of the lipid extract. N = 9. Error bars indicate standard error of mean (SEM). Student's t-test was performed. ** = P-value < 0.01. Please click here to view a larger version of this figure.
Table 1: Table showing the set-up of standards for the organic phosphate assay. The amount of stock solution (concentration = 7.34 mM) and water to be mixed to make each dilution is shown. Please click here to download this Table.
Table 2: List of PA in DIMS targeted approach. The first two columns show the MASS = mass of species and CHEMSEC = the chemical formula. The third column NAME = name of the PA molecule. Please click here to download this Table.
Table 3: A screenshot of the output in .csv format from the Lipidxplorer software. Column 1 – mass of PA molecule; column 2 – the chemical formula of PA molecule; column 3 – error is in ppm; column 4 – name of the PA molecule, columns 5 to 15- intensity of individual test samples. Please click here to download this Table.
Table 4: The experimental parameters used for the analysis of all the PA molecular species in NPLC-MRM-EPI method. MOLECULAR ID – molecular name of PA, Q1 MASS (Da) – mass selected in quadrupole 1, Q3 MASS (Da) – mass selected in quadrupole 3, Retention time – retention time of the PA molecules, Dwell Time – dwell time in msec, CE – collision energy used for MS/MS fragmentation, DP – declustering potential, EP – exit potential, CXP – collision cell exit potential. Please click here to download this Table.
Table 5: Screenshot of neutral mass. TMSD derivatized mass and the DAG fragments of some representative PA molecules. Column A shows the lipid class, column B shows the total number of carbon in two fatty acyl chains, column C shows the total number of double bonds in two fatty acyl chains, column D shows the chemical formula of the lipid molecules, column E shows the neutral mass of the lipid molecule, column F shows the methylated mass after derivatization using TMSD, column G shows the protonated mass of the methylated lipid molecule, and column H shows the DAG fragment arise after collision induced dissociation of the protonated methylated lipid molecule. Please click here to download this Table.
