Most complex eukaryotic proteins undergo elaborate posttranslational modifications after expression, requiring highly assisted protein folding and co-factors to be functional1. Producing large amounts of soluble human protein in a bacterial host remains a significant challenge due to high costs and the lack of robust expression and purification methods, even for smaller-scale laboratory experiments2,3. MMPs, human endopeptidases with large molecular weight, are usually expressed as insoluble inclusion bodies when expressed in E. coli. Extraction of soluble human MMPs often leads to a laborious, time-consuming solubilization and refolding process4.
MMPs have critical roles in both physiological and pathogenic processes. Human MMPs are a family of 23 zinc endopeptidases, categorized by structure and substrate specificity, and differentially expressed in spite of a highly conserved catalytic domain5,6. MMPs are secreted as inactive zymogens, regulated via posttranslational activation and their endogenous inhibitors, tissue inhibitors of metalloproteinases (TIMPs)7,8,9,10. Though initially recognized for their role in ECM turnover, MMPs have also been implicated in development, morphogenesis, tissue repair, and remodeling8. Dysregulation of MMPs has been notably linked to cancer along with neurodegenerative, cardiovascular, and fibrotic diseases, among other illnesses5,7.
The development of robust large-scale MMP production methods is critical to ensure the success of future studies of MMP mechanisms through biochemical and cell-based assays. Various MMPs have been previously expressed in bacteria11, including Hisx6-tagged MMPs, without altering MMP activity12,13,14,15. However, these methods include tedious, long steps that might be difficult to replicate.
Mammalian cells can also be used to express many different human proteins while ensuring the proper posttranslational modifications16. Although the mammalian expression system is an ideal choice to produce recombinant human proteins with proper post-translational modifications, the main disadvantages of this method are initial low yields, costly growth media and reagents, long timelines to reach stable expression lines, and risk of contamination with other species such as fungi or bacteria2,11. Moreover, MMP production in mammalian cell lines yields impurities from associated cellular proteins such as TIMPs or fibronectins11. Unlike the slow cell growth observed in mammalian cells, the bacterial expression system offers large-scale protein production in a short period along with simpler media and growth requirements. However, due to the lack of other associated cellular proteins (i.e., TIMPs) in bacterial expression systems, active MMPs at higher concentrations are subject to degradation through autoproteolysis, resulting in poor MMP yield17.
This paper describes a detailed method for bacterial expression, purification, and activation of recombinant Hisx6-pro-MMP-3cd using E. coli as an expression host due to its affordability, simplicity, and success in producing higher yields of MMPs2,3,18. Since E. coli lacks the protein folding machinery and posttranslational processing required for recombinant MMPs and other complex proteins, many E. coli strains have been engineered to overcome these limitations, making E. coli a more suitable host for expression of recombinant human MMP-3cd,19,20. For instance, the R2DP strain used in this study enhances eukaryotic expression by supplying a chloramphenicol-resistant plasmid containing codons rarely used in E. coli.
As described in this protocol, after overexpression of relatively pure inclusion bodies from the pET-3a vector (Figure 1) in R2DP cells, Hisx6-pro-MMP-3 catalytic domain (MMP-3cd) proteins are extracted and denatured4. Hisx6-pro-MMP-3cd3,19 was purified using affinity tag chromatography. Upon refolding and dialysis, the pro-MMP-3cd (zymogen) was activated by 4-aminophenylmercuric acetate (APMA), and SDS-PAGE analysis is used to evaluate yields and the need for further purification5,21. This protocol describes expression, purification, and activation of soluble MMP-3cd as an example. However, it may be also used as a guide for expression of other MMPs and human proteases with similar expression, and activation mechanisms (Figure 2). For other proteins other than MMP-3cd, the reader is advised to determine optimal buffer compositions and methods for their target protein before attempting this protocol.
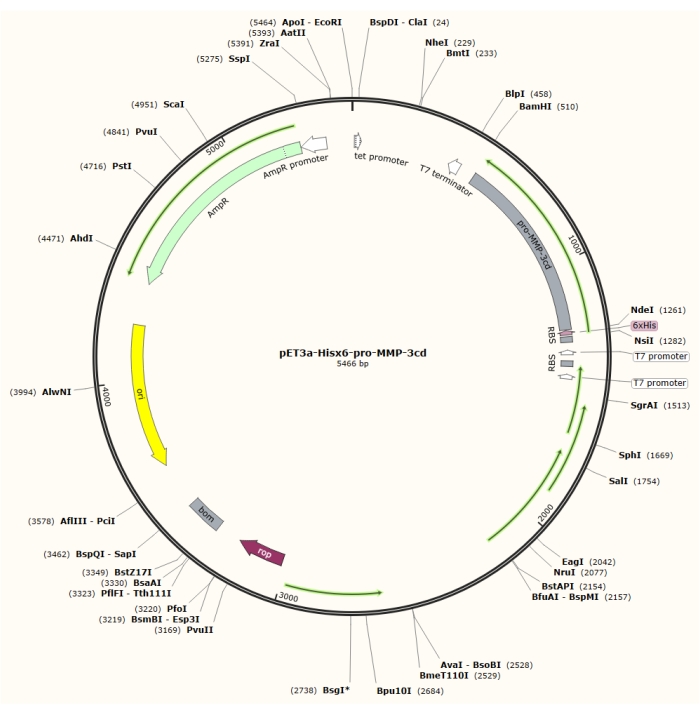
Figure 1: Plasmid map of the pET-3a-Hisx6-pro-MMP-3cd plasmid. The pET-3a vector includes an ampicillin resistance gene. An N-terminal Hisx6-tag sequence is cloned into the pET-3a-based vector, including pro-MMP-3cd, to yield the pET-3a-Hisx6-pro-MMP-3cd construct under control of T7 promoter between BamHI and NdeI restriction sites. Please click here to view a larger version of this figure.
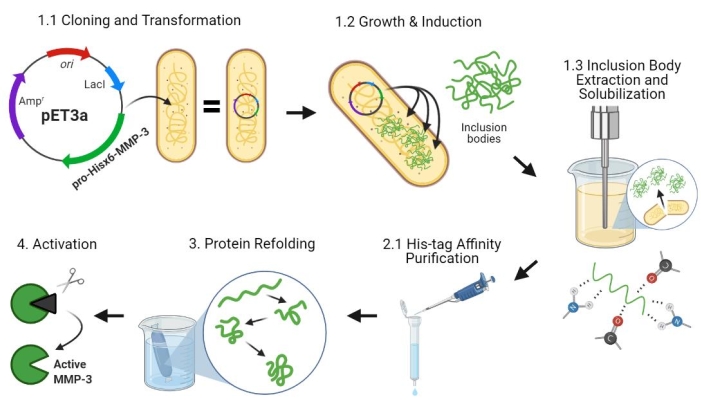
Figure 2: Bacterial expression of pro-MMP-3cd, purification, refolding, and activation. 1.1: pET-3a-Hisx6-pro-MMP-3cd plasmid was transformed into BL21(DE3) or R2DP Cells. 1.2: Pro-MMP-3cd protein expression was induced using IPTG. 1.3: Chemical lysis and sonication are used to extract Hisx6-pro-MMP-3cd proteins that are mainly insoluble and found in the inclusion bodies. Urea was used to denature and solubilize protein from inclusion bodies. 2.1. Denatured Hisx6-pro-MMP-3cd protein was purified via affinity chromatography purification. 3. The eluted Hisx6-pro-MMP-3cd was slowly refolded during dialysis through gradual removal of urea from the buffer. 4. Finally, refolded MMP-3cd protein was activated using APMA by removing the N-terminal pro-peptide domain. APMA is later removed from the solution through desalting. The numbers correspond to protocol sections describing these steps. Abbreviations: MMP-3cd = Matrix metalloproteinase-3 catalytic domain; APMA = 4-aminophenylmercuric acetate. Please click here to view a larger version of this figure.
When running samples on SDS-PAGE, because the protein is expressed in the form of insoluble inclusion bodies, the lysed and sonicated fractions should contain little to no Hisx6-pro-MMP-3cd extract, as the protein has not yet been resolubilized in urea. Figure 3 compares the His-tag purification elution fractions of Hisx6-pro-MMP-3cd from BL21(DE3) cells and R2DP cells. Elution fractions were pooled separately for both BL21(DE3) and R2DP cells before dialysis. Fractions from each step were run after the proteins were desalted (Figure 4). All Hisx6-pro-MMP-3cd samples display a band at approximately 30 kDa, and active MMP-3cd displays a band at approximately 20 kDa upon removal of the His-tag and pro-domain (Figure 4 and Supplemental Figure S1).
The total yield of protein in mg/L of E. coli culture was determined after purification and desalting of BL21(DE3) and R2DP cultures (Table 1). Using R2DP cells yields substantially higher levels of MMP expression. Whereas regular BL21(DE3) cells yielded only 3.5 mg of purified Hisx6-pro-MMP-3cd per liter of culture, R2DP cells produced 45 mg /L culture. Similarly, yields of functional, desalted MMP-3cd increased from 0.13 mg/L culture to 6.2 mg/L culture for BL21(DE3) and R2DP cells, respectively. Human pro-MMP-3cd overwhelms the cellular machinery of the standard BL21(DE3) strain because of its size (approximately 30 kDa) and the elaborate posttranslational modifications required that are exclusive to eukaryotes. The R2DP strains are BL21(DE3) derivatives, designed to enhance the expression of eukaryotic proteins. The R2DP strain carries tRNAs for AGA, AGG, AUA, CUA, GGA, CCC, and CGG, which are rarely used in E.coli but abundant in the pro-MMP-3cd DNA sequence. This is potentially a key factor in the increased levels of protein expression observed in R2DP cells.
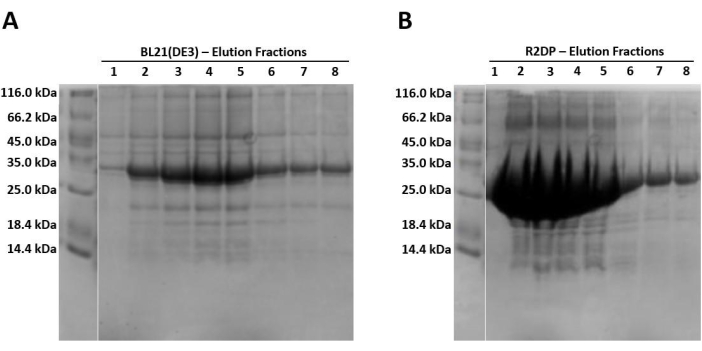
Figure 3: SDS-PAGE gel analysis of Hisx6-pro-MMP-3cd expression in BL21(DE3) and R2DP cells. (A) The first eight elution fractions of Hisx6-pro-MMP-3cd in BL21(DE3) cells. (B) The first eight elution fractions of Hisx6-pro-MMP-3cd in R2DP cells. Following extraction and solubilization of MMP inclusion bodies in urea, Hisx6-pro-MMP-3cd samples were purified through Ni-NTA chromatography column using batch-gravity flow. Gels are truncated to show only the elution fractions. Initially, due to high concentrations of protein, fractions 1-5 are 1 mL. Later fractions (6-8) are between 5 and 8 mL each. The Hisx6-pro-MMP-3cd band is observed at ~30 kDa. Abbreviations: MMP-3cd = Matrix metalloproteinase-3 catalytic domain; SDS-PAGE = sodium dodecylsulfate polyacrylamide gel electrophoresis; Ni-NTA = nickel-nitrilotriacetic acid; FT = flowthrough. Please click here to view a larger version of this figure.
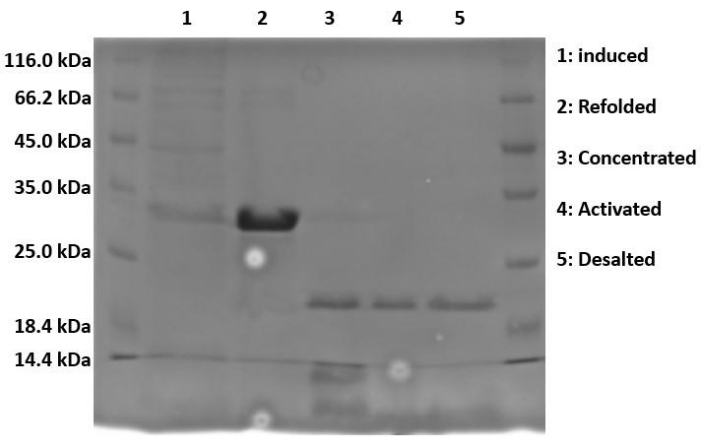
Figure 4: Proteolytic cleavage of His-tag and pro-domain upon activation of MMP-3cd. The induced, refolded, concentrated, activated, and desalted fractions of MMP-3cd in R2DP cells are shown. After dialysis, Hisx6-pro-MMP-3cd is concentrated and activated using APMA. Upon activation, the molecular weight of the activated MMP-3cd band is approximately 20 kDa, as opposed to the His-tagged zymogen, which remains at ~30 kDa. Impurities are removed in the activation and desalting stages. Abbreviations: MMP-3cd = Matrix metalloproteinase-3 catalytic domain; APMA = 4-aminophenylmercuric acetate. Please click here to view a larger version of this figure.
| Stages | BL21(DE3) cells | R2DP cells | ||||
| Volume (mL) | Concentration (mg/mL) | Yield (mg/L culture) | Volume (mL) | Concentration (mg/mL) | Yield (mg/L culture) | |
| Purification | 23 | 0.30 | 3.5 | 42 | 2.1 | 45 |
| Desalting | 1.5 | 0.17 | 0.13 | 72 | 0.17 | 6.2 |
Table 1: Table of volumes and concentrations across stages of MMP-3cd purification. Hisx6-pro-MMP-3cd was expressed either in 2 L of a culture of BL21(DE3) or in 2 L of R2DP cells. Volume, concentration, and yield (mg per liter) culture are reported for BL21(DE3) and R2DP cells. Yield (mg per liter of culture) was obtained by dividing the total yield of protein (mg) obtained by volume of culture, which was 2 L for both the BL21(DE3) and R2DP cases. Protein amounts yields are reported for two stages: Hisx6-pro-MMP-3cd following Hisx6-tag purification and active MMP-3cd after desalting.
Supplemental Figure S1: Sequences of T7 primers and MMP-3cd protein before and after activation. Please click here to download this File.
