Most of the excitatory synapses in the central nervous system are located on dendritic spines – small protrusions of a neuronal membrane. These protrusions form confined biochemical compartments that control intracellular signal transduction. Structural plasticity of dendritic spines and synapses is closely related to the functional changes in synaptic efficacy that underlie such important processes as learning and memory1,2. It is important to note that electron microscopy (EM) is the only technique that allows to determine if a dendritic spine has a presynaptic input. EM resolution is also needed to study ultrastructural details such as shape of a postsynaptic density (PSD), representing a postsynaptic part of a synapse, or dimensions of a dendritic spine, as well as the size and shape of an axonal bouton. Additionally, with EM it is possible to visualize synapses and their surroundings.
Thanks to advances in imaging and computing technologies it is possible to reconstruct entire neural circuits. Volume electron microscopy techniques, such as serial section transmission electron microscopy (ssTEM), serial block-face scanning electron microscopy (SBEM), and focused ion beam scanning electron microscopy (FIB-SEM) are commonly used for neuronal circuit reconstructions3.
In our studies, the SBEM method is successfully employed to investigate the structural plasticity of dendritic spines and PSDs in samples of the mouse hippocampus and organotypic brain slices 4,5. The SBEM is based on the installation of a miniature ultramicrotome inside the scanning electron microscope chamber6,7,8,9. The top of the sample block is imaged, and then the sample is cut at a specified depth by the ultramicrotome, revealing a new block-face, which is again imaged and then the process is repeated8. As a result, only the image of a block-face is left while the slice which has been cut is lost as debris. That is why SBEM is called a destructive technique, meaning it is not possible to image the same place again. However, the advantage of the destructive on-block methods is that they do not suffer from warping problems and section loss that can significantly affect the data quality and the data analysis3. Moreover, SBEM gives the possibility to image a relatively large field of view ( > 0.5 mm × 0.5 mm) at high resolution3.
To employ SBEM, samples have to be prepared according to a dedicated, highly contrasting protocol due to the backscattered electron detector used for acquiring images. We show here how to perform sample preparation according to the protocol based on a procedure developed by Deerinck10 (National Center for Microscopy and Imaging Research (NCMIR) method), using reduced osmium-thiocarbohydrazide-osmium (rOTO) stains developed in the 1980s8,11. In addition, we introduce a two-step fixation approach, with mild fixation perfusion that allows to use the same brain both for immunofluorescence studies with light microscopy and SBEM.
In the protocol a mouse brain is primarily fixed with a mild fixative, and then cut into halves, and one hemisphere is postfixed and prepared for immunofluorescence (IF), while the other for EM studies (Figure 1).

Figure 1. Schematic representation of the workflow for the dendritic spines preparation for the analysis with SBEM. Mice were sacrificed and perfused with a mild primary fixative. The brain was cut into halves, and one hemisphere was postfixed with immunofluorescence (IF)-dedicated fixative, cryoprotected, sliced using a cryostat and processed for IF studies, while the other hemisphere was postfixed with EM fixative, sliced with the vibratome and prepared for EM studies. Brain slices for SBEM studies were contrasted, flat embedded in resin, then a CA1 region of the hippocampus was mounted to the pin, and imaged with SBEM (Figure 1). The part of the protocol highlighted in a yellow box was featured in the video. Please click here to view a larger version of this figure.
Using the method described above high contrast, good resolution images of the mouse brain tissue can be obtained. A large field of view provided by the SBEM technique facilitates precise selection of the region of interest. The large image of the CA1 region of the hippocampus was taken to measure the length of stratum radiatum (SR) (Figure 2A) and to set the imaging precisely in the center (Figure 2B). Next, the stack of images was acquired and the objects of interest, such as dendrites, dendritic spines, postsynaptic densities of the synapses were segmented (Figure 2C,D).
The use of the SBEM technique gives the possibility to analyze a relatively large volume of tissue and to recognize rare events such as a multi-innervated dendritic spine (Figure 2E) – a particular type of synapses that is characterized by several presynaptic terminals contacting the same postsynaptic spine21.

Figure 2. Dendritic spines in mouse hippocampus CA1 region. Representative image of mouse hippocampal CA1 region. Stratum radiatum (SR) is shown with a white arrow (A). The region in the middle of SR was chosen for imaging (B). Reconstruction of a piece of dendrite with dendritic spines (C). Reconstruction of dendritic spines in a volume of tissue (D). Reconstruction of a multi-innervated dendritic spine (E). Two presynaptic terminals contacting the same postsynaptic spine are shown (in magenta and green). Please click here to view a larger version of this figure.
The presented protocol for SBEM sample preparation allowed us to obtain high-quality images of the brain tissue with heavily contrasted membranes, which enables easier segmentation and reconstruction of membrane surrounded structures. The standard fixation with the higher concentration of PFA (Figure 3B) and the two-step fixation approach resulted in similar tissue morphology (Figure 3A).

Figure 3. Morphological details. With the use of the protocol presented here good quality images showing the morphology of synaptic vesicles, PSD and mitochondria were obtained. Brain tissue fixed with two-step approach (A) or with a standard fixative (B). Scale bar 2 µm. Please click here to view a larger version of this figure.
The results are reproducible, meaning that every specimen prepared using this protocol, not only brain tissue but also the monolayer of cultured cells, was well contrasted.
The use of mild primary fixation offers the opportunity to use the tissue also for immunofluorescence studies involving a light microscope. In the case of the PSD-95 antibody, we did not observe the difference in immunostaining results between the samples fixed with two-step fixation (mild primary and then secondary with a traditional fixative) and the one obtained using traditional fixation with 4% PFA only (Figure 4A and 4B, respectively).

Figure 4. Comparison of PSD-95 immunofluorescence in samples fixed with various protocols. Representative microphotographs of PSD-95 immunostaining in the stratum radiatum layer of CA1 area in the dorsal hippocampus. (A) Confocal image after mild primary fixation followed by postfixation with a standard fixative. (B) Confocal image after standard, stronger fixation. Scale bars: 5 µm. Please click here to view a larger version of this figure.
| Anesthetic: | |||
| Ketamine/xylazine mixture (Ketamina/Sedazin) | Biowet Pulawy, Pulawy, Poland | ||
| Sodium pentobarbital (Morbital) | Biowet Pulawy, Pulawy, Poland | ||
| Fixatives: | |||
| Glutaraldehyde (GA) | Sigma-Aldrich,St. Louis, MI, USA | G5882 | Grade I, 25% in H2O, specially purified for use as an electron microscopy fixative |
| Hydrochloric acid (HCl) | POCH, Gliwice, Poland | 575283115 | pure p.a. |
| Paraformaldehyde (PFA) | Sigma-Aldrich,St. Louis, MI, USA | 441244 | prilled, 95% |
| Phosphate buffered saline (PBS), pH 7.4 | Sigma-Aldrich,St. Louis, MI, USA | P4417-50TAB | tablets |
| Sodium hydroxide (NaOH) | Sigma-Aldrich,St. Louis, MI, USA | S5881 | reagent grade, 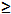 98%, pellets (anhydrous) 98%, pellets (anhydrous) |
| Sodium phosphate dibasic (Na2HPO4) | Sigma-Aldrich,St. Louis, MI, USA | S3264 | |
| Sodium phosphate monobasic (NaH2PO4) | Sigma-Aldrich,St. Louis, MI, USA | S3139 | |
| Perfusion: | |||
| Large blunt/blunt curved scissors (~14.5 cm) | Fine Science Tools, Foster City, CA, USA | 14519-14 | |
| Micro-spatula (double 2" flat ends, one rounded, one tapered to 1/8") | Fine Science Tools, Foster City, CA, USA | 10091-12 | |
| Needle tip, 15 GA, blunt (perfusion needle) | KD Medical GmbH Hospital Products, Berlin, Germany | KD-FINE 900413 | 1.80 x 40 mm |
| Pair of fine (Graefe) tweezers | Fine Science Tools, Foster City, CA, USA | 11050-10 | |
| Perfusion pump | Lead Fluid | BQ80S | |
| Plastic vials | Profilab, Warsaw, Poland | 534.02 | plastic vials with blue cap for tissue storage, 20 ml, 31 x 48 mm |
| Straight iris scissors (~9 cm) | Fine Science Tools, Foster City, CA, USA | 14058-11 | |
| Brain slices preparation for EM: | |||
| 12-well plate | NEST, Rahway, NJ, USA | 712001 | |
| Cyanoacrylic glue | Fenedur, Montevideo, Uruguay | ||
| Glass vials | Electron Microscopy Sciences, Hatfield, PA, USA | 72632 | 20 ml Scintillation Vial, a pack of 100 |
| Pasteur pipette | VWR, Radnor, PA, USA | 612-4545 | LDPE, disposable, 7.5 ml |
| Razor blade | Wilkinson Sword, London, UK | Classic double edge safety razor blades | |
| Scalpel blade | Swann-Morton, Sheffield, UK | No. 20 | |
| Vibratome | Leica Microsystems, Vienna, Austria | Leica VT1000 S | |
| Brain slices preparation for IF: | |||
| 96-well plate | NEST, Rahway, NJ, USA | 701101 | |
| Criostat | Leica Microsystems, Vienna, Austria | Leica CM 1950 | |
| Ethylene glycol | Bioshop, Burlington, Canada | ETH001 | |
| Low-profile disposable blade 819 | Leica Biosystems Inc., USA | 14035838925 | |
| Scalpel blade | Swann-Morton, Sheffield, UK | No. 20 | |
| Sodium azide (NaN3) | POCH, Gliwice, Poland | 792770426 | |
| Sucrose | POCH, Gliwice, Poland | 772090110 | |
| Tissue freezing medium for cryosectioning, OCT-Compound | Leica Biosystems, Switzerland | 14020108926 | |
| Immunostaining: | |||
| 24-well plate | NEST, Rahway, NJ, USA | 702001 | |
| Anti-Post Synaptic Density Protein 95 Antibody | Merck-Millipore, Burlington, MA, USA | MAB1598 | |
| Confocal microscope | Zeiss, Göttingen, Germany | Zeiss Spinning Disc microscope (63 × oil objective, NA 1.4, pixel size 0.13 µm × 0.13 µm) | |
| Cover slide | Menzel Glaser, Braunschweig, Germany | B-1231 | 24 x 60 mm |
| Donkey anti-Mouse IgG (H+L) Highly Cross-Adsorbed Secondary Antibody, Alexa Fluor 555 | Invitrogen, Carlsbad, CA, USA | A31570 | |
| Fluoromount-G Mounting Medium, with DAPI | Invitrogen, Carlsbad, CA, USA | 00-4959-52 | |
| Microscope slide | Thermo Scientific, Waltham, MA, USA | AGAA00008 | SuperFrost |
| Normal donkey serum (NDS) | Jackson ImmunoResearch Laboratories, West Grove, PA, USA | 017-000-121 | |
| Shaker | JWElectronic, Warsaw, Poland | KL-942 | |
| TritonT X-100 Reagent Grade | Bioshop, Burlington, Canada | TRX506 | |
| Electron microsocpy sample preparation | |||
| Potassium hexacyanoferrate(II) trihydrate | POCH, Gliwice, Poland | 746980113 | |
| Aclar 33C Film | Electron Microscopy Sciences, Hatfield, PA, USA | 50425 | Fluoropolymer Film embedding sheet |
| DMP-30, 2,4,6-Tris(dimethylaminomethyl)phenol | Sigma-Aldrich,St. Louis, MI, USA | T58203 | Epoxy embedding medium accelerator |
| Durcupan ACM single component A, M | Sigma-Aldrich,St. Louis, MI, USA | 44611 | Durcupan ACM single component A, M epoxy resin |
| Durcupan ACM single component B | Sigma-Aldrich,St. Louis, MI, USA | 44612 | Durcupan ACM single component B, hardener 964 |
| Durcupan ACM single component D | Sigma-Aldrich,St. Louis, MI, USA | 44614 | Durcupan ACM single component D , plasticizer |
| Ethyl alcohol absolute | POCH, Gliwice, Poland | 64-17-5 | Ethyl alcohol absolute 99.8 % pure P.A.-BASIC |
| Genlab laboratory oven | Wolflabs, York, UK | Mino/18/SS | Oven Genlab MINO/18/SS 18l volume, no fan circulation, no digital display, standard temperature gradient, standard recovery rate, no timer, 250°C maximum temperature, 240V electrical supply |
| L-Aspartic acid | Sigma-Aldrich,St. Louis, MI, USA | A-9256 | reagent grade, 98% (HPLC) |
| Lead (II) nitrate | Sigma-Aldrich,St. Louis, MI, USA | 467790 | 99.95% trace metals basis |
| Osmium tetroxide | Sigma-Aldrich,St. Louis, MI, USA | 75632 | for electron microscopy, 4% in H2O |
| pH meter | Elmetron, Zabrze, Poland | CP-5-5 | |
| Rotator | BioSan, Józefów, Poland | Multi Bio RS-24 | rotator Multi Bio RS-24 |
| Sodium hydroxide (NaOH) | Sigma-Aldrich,St. Louis, MI, USA | S5881 | reagent grade, 98%, pellets (anhydrous) |
| Sunflower mini shaker | Grant bio, Shepreth,UK | PD-3D | |
| Syringe filter | Millipore, Burlington, MA, USA | SLGP033NB | 0,22 µm pore size |
| Thiocarbohydrazide | Sigma-Aldrich,St. Louis, MI, USA | 88535 | purum p.a., for electron microscopy, 99.0% (N) |
| Uranyl acetate | Serva, Heidelberg, Germany | 77870 | Uranyl acetate·2H2O, research grade |
| Water bath | WSL, Swietochlowice, Poland | LWT | |
| Specimen mounting for SBEM | |||
| 96-well culture plate | VWR, Radnor, PA, USA | 734-2782 | 96-well plates, round bottom, non treated |
| AM Gatan 3View stub handling tweezers | Micro to Nano, Haarlem, Netherlands Netherlands |
50-001521 | |
| Binocular | OPTA-TECH, Warsaw, Poland | X2000 | |
| Conductive glue | Chemtronics, Georgia, USA | CW2400 | conductive eopxy |
| Gatan 3View sample pin stubs | Micro to Nano, Haarlem, Netherlands Netherlands |
10-006003 | |
| Parafilm | Sigma-Aldrich,St. Louis, MI, USA | P7793 | roll size 20 in. × 50 ft |
| Pelco conductive silver paint | Ted Pella, Redding, CA, USA | 16062-15 | PELCO® Conductive Silver Paint, 15g |
| Razor blades double edge | Electron Microscopy Sciences, Hatfield, PA, USA | 72000 | Stainless Steel "PTFE" coated. PERSONNA brand .004" thick, wrapped individually, 250 blades in a box. |
| Scanning Electron Microscope | Zeiss, Oberkochen, Germany | Sigma VP with Gatan 3View2 chamber, acceleration voltage 2.5 kV, variable pressure 5 Pa, aperture 20 µm, dwell time 6 µs, slice thickness 60 nm, magnification 15 000 x, image resolution 2048 x 2048 pixels, pixel size 7.3 nm | |
| trim 90° diamond knife | Diatome Ltd., Nidau, Switzerland | DTB90 | |
| Ultramicrotome | Leica Microsystems, Vienna, Austria | Leica ultracutR | |
| Software | webpage | tutorials | |
| FijiJ | https://fiji.sc/ | ||
| Microscopy Image Browser | http://mib.helsinki.fi/ | http://mib.helsinki.fi/tutorials.html | |
| Reconstruct | https://synapseweb.clm.utexas.edu/software-0 | https://synapseweb.clm.utexas.edu/software-0) | |
| Animals | |||
| Mice | Adult 3-month old and 20±1 month old female Thy1-GFP(M) mice (Thy1-GFP +/-) (Feng et al.,2000) which express GFP in a sparsely distributed population of glutamatergic neurons. Animals were bred as heterozygotes with the C57BL/6J background in the Animal House of the Nencki Institute of Experimental Biology. |
