NOTE: This protocol spans 2 days with a minimum period of 4-5 days for viral transduction between experimental days (Figure 1A). Animal experiments are performed in accordance with the Canadian Council on Animal Care Guidelines for Use of Animals in Research and Laboratory Animal Care under protocols approved by the Laboratory of Animal Services Animal Use and Care Committee at the Hospital for Sick Children (Toronto, Canada).
1. Preparations for the neonatal AAV injections and imaging experiments
- Select a Cre mouse line to label the retinal cell populations of interest. Confirm Cre recombinase expression at the time of AAV injections by crossing to a Cre transgenic reporter or through immunostaining with a Cre antibody.
- Breed transgenic Cre mice (8-16 weeks old) to generate Cre-positive neonatal animals.
NOTE: For this demonstration, ChAT-IRES-Cre knock-in mice were used to target the cholinergic Starburst amacrine cells. - Obtain recombinase-dependent AAV virus encoding fluorescent protein(s). For optimal labeling of fine processes, select vectors that express modified XFPs targeting the plasma membrane.
NOTE: This study used the Brainbow virus (BBV), AAV-EF1a-BbTagBY and AAV9-EF1a-BbChT (see Table of Materials), which provide the option for multicolour labeling (Figure 1B). Farnesylated enhanced yellow fluorescent protein (eYFP) and monomeric Cherry (mCherry) expression produced the strongest fluorescent signals for live imaging. A single BBV can be used to image individual arbors, while co-injection of BBVs can be used to label more cells or neighboring arbors with distinct fluorophores. If using multiple fluorescent proteins, ensure minimal emission spectrum overlap (Figure 1C). Microscope acquisition parameters must be adjusted to capture distinct XFP signals. - Prepare ∼3-5 µL aliquots of AAVs in individual low-binding tubes for single-use stocks to avoid freeze/thaw cycles. Store at -80 °C.
- Prepare borosilicate glass micropipettes with very fine tips using a micropipette puller.
NOTE: Puller settings vary depending on filament and puller being used; see Figure 2A for final tip size and shape. Typical droplet diameters are 600 µm. - Obtain a microinjection system and associated tubing. Connect the microinjector to a pressurized air port.
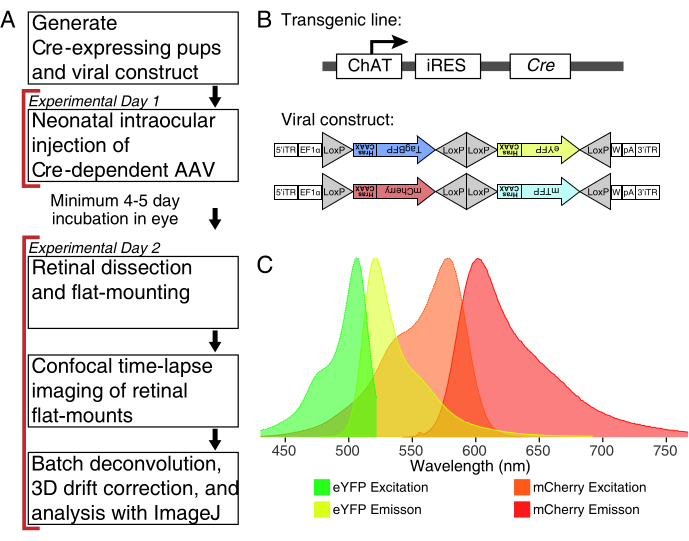
Figure 1: Experimental overview. (A) This protocol spans 2 days of experiments with a minimum of 4-5-day infection period between experimental days. Intraocular injections are performed on neonatal mice no older than postnatal day 3. Retinas are then dissected, flat-mounted, and live-imaged to capture the desired developmental window. Labelled cells can be imaged any time after the required 4-5 days needed for viral expression, as there are no apparent effects of prolonged AAV expression on dendrite morphology. (B) Cre-dependent Brainbow AAV vectors (BBV) are injected into animals expressing Cre31. In this study, ChAT-Cre knock-in mice were used to drive Cre recombination in starburst amacrine cells. The two BBVs encode modified eYFP or tagBFP, or mCherry and mTFP, which terminate in a CAAX motif that is sequentially farnesylated for membrane localization. Lox sites are depicted with triangles. The vectors express either farnesylated XFPs in a stochastic and combinatorial manner dependent on Cre recombination. The EF1α promoter includes regulatory elements from the elongation factor 1α gene. W represents elements from the woodchuck hepatitis virus posttranscriptional regulatory element, and pA indicates the polyadenylation sequence. (C) The excitation and emission spectra of mCherry and eYFP, the BBV fluorophores imaged in this study. When live-imaging multiple fluorescent proteins, the detection parameters must be arranged to adequately separate emission spectra into distinct channels. Abbreviations: AAV = adeno-associated virus; BBV = Brainbow AAV; ChAT = choline acetyltransferase; iRES = internal ribosome entry site; eYFP = enhanced yellow fluorescent protein; iTR = inverted terminal repeat; tagBP = Tag-blue fluorescent protein; mCherry = monomeric Cherry; mTFP = teal fluorescent protein; XFP = any fluorescent protein; EF1α= elongation factor 1 alpha. Please click here to view a larger version of this figure.
2. Intravitreal injections of AAVs in neonatal mice
- Thaw an AAV aliquot on ice. Prepare a ~1:4 AAV dilution using sterile saline or phosphate-buffered saline. Prepare ~0.5 µL of AAV dilution per animal (~0.25 µL per eye) in case the micropipette breaks, and a new pipette needs to be filled. Store the remaining undiluted AAV at 4 °C, and use within 2 weeks.
NOTE: In this experiment, the supplier AAV concentration was ~1-2 × 1013 genome content (GC)/mL and diluted to a final concentration of 4 × 1012 GC/mL. Optimize the viral concentration to obtain the desired labeling density. - To visualize the injections, add approximately 1 µL of 0.02% Fast Green FCF dye solution for every 15 µL of AAV dilution to color the solution blue.
- Transfer a mouse cage with dam and newborn litter (postnatal day (P) 0.5-3) to a procedure room with microinjection equipment. Keep the animals in the same cage with nests and bedding to minimize maternal stress and rejection of pups.
- Sterilize the injection area with 70% ethanol, and place sterile diaper pads on bench surfaces. Prepare a warm platform (e.g., a heating pad) for recovery from hypothermia-induced anesthesia.
- Backfill the micropipette with the AAV dilution using a microsyringe. Under a stereomicroscope, break the micropipette tip with a 30 G needle to unseal the tip.
NOTE: Figure 2A shows the backfilled tip-both sealed (top) and unsealed (middle). - Anesthetize neonatal mice by hypothermia by placing 1-2 animals on ice. Place animals onto a latex glove to protect skin from direct contact with ice. Once the animal no longer moves in response to paw pinch (~2 min), place the animal under a stereomicroscope. If desired, tattoo paw pads with tattoo ink and 30 G needle, and collect tail clippings for DNA isolationto identify animals by genotyping.
NOTE: Appropriate monitoring of depth of anesthesia must occur throughout the procedure. - Swab the skin overlying the eyes with 70% ethanol. Use a 30 G needle to open the fused eyelid (Figure 2B). Apply light pressure with the fingers to open the eye, and poke a small hole through the cornea at the cornea-sclera junction (Figure 2C).
- Insert the glass micropipette into the hole, and press the microinjector foot pedal 2-4 times to inject the AAV into the intravitreal space. Slowly remove the micropipette, and confirm AAV injection by visualizing blue dye through the pupil (Figure 2D).
NOTE: With an ejection pressure of 6-8 psi and pulse time of 600-800 ms, a 600 µm diameter droplet is ejected (Figure 2A, bottom). Approximately 0.23-0.45 µL of AAV solution is injected per eye. Blue solution outside the eye indicates that the AAV was not injected into the eye. Blue solution leaking from the injection site indicates that the AAV may have leaked out, reducing transfection efficiency. - Gently press the eyelids together to re-seal, and place the pup onto a heated pad. Once the animals recover a pinkish color and are responsive, gently transfer them back to the housing cage.
NOTE: Ensure appropriate precautions are taken to avoid too rapid re-warming of the pups. - Repeat the injection procedure for the remaining animals in the litter. Allow a minimum of 4-5 days for viral transduction before imaging the retina.
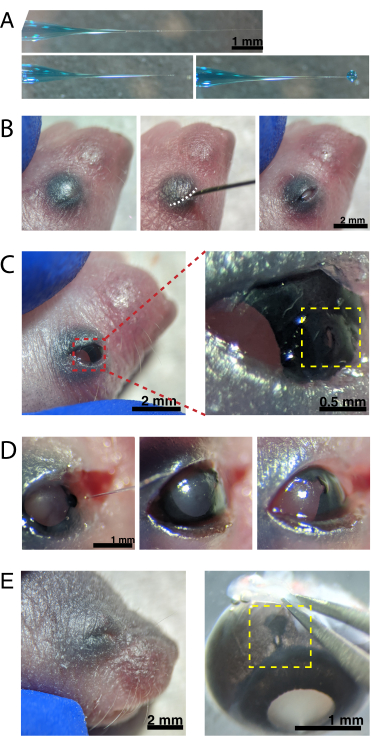
Figure 2: Neonatal intraocular injections. (A) Backfilled micropipette shows the shape of the pipette tip when sealed (top) and after the tip is unsealed (bottom, left). Pico-injection pressure of 6-8 psi and pulse time of 600-800 ms produces a 600 µm diameter droplet (bottom, right). Scale bar = 1 mm.(B) Anesthetized P0 pup under 16x magnification. The fused eyelid junction (white) is slit open using a sharp 30 G needle. Scale bar = 2 mm. (C) Light pressure applied to the eye exposes the cornea; scale bar = 2 mm. Zoomed section (red) shows the small hole at the cornea-sclera junction (yellow) created with a 30 G needle; scale bar = 0.5 mm. (D) Withdrawal of the pipette tip after injection (left). Fast Green dye in the AAV solution appears as blue-grey through the pupil (middle), as compared to the light pupil color before injection (right). Scale bar = 1 mm. (E) After 4 days, the eyelid has healed and fused shut (left); scale bar = 2 mm. Upon enucleation, the healed injection site is visible (yellow); scale bar = 1 mm. Note the location of the injection site at the border between the cornea and sclera. Please click here to view a larger version of this figure.
3. Retinal dissections for imaging experiment
- Prepare retinal aCSF (119 mM NaCl, 2.5 mM KCl, 1.3 mM MgCl2·6H2O, 2.5 mM CaCl2·2H2O, 1 mM NaH2PO4, 11 mM glucose, and 20 mM 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES free acid)35. If desired, prepare and freeze 10x solution; thaw and dilute to 1x as needed.
- Oxygenate the retinal aCSF by bubbling with carbogen (95% O2, 5% CO2) for a minimum of 15 min. Adjust the pH to 7.4. Keep in a sealed container until use to ensure that the aCSF remains oxygenated.
NOTE: Retinas require a high concentration of O2. It is important to keep aCSF oxygenated throughout the experiment. - Embed a 60 mm diameter Petri dish into an ice tray (fashion an ice tray out of lab plastic, e.g., a pipette box lid), and place it under a stereomicroscope. Fill the Petri dish with oxygenated retinal aCSF.
- Euthanize mice P9 and younger by decapitation. Euthanize mice P10 and older by isoflurane induction, or an alternate method approved under the animal protocol, followed by decapitation.
- If the eyelids are sealed, cut the eyelid flap to expose the eye. Use forceps to enucleate the eyes, and transfer them into the cold retinal aCSF from step 3.3.
- To dissect the retinal cup, under a stereomicroscope (magnification 25x), stabilize the eye by clasping the optic nerve using a Dumont #5 forcep (Figure 3A).
- Poke a hole in the center of the cornea with a 30 G needle, and then insert one tip of the microscissors into the hole to make an incision from the hole to the end of the cornea. Repeat to make 4 slices in the cardinal directions, creating 4 "flaps" (Figure 3A).
- With two Dumont #5 forceps, grasp and pull the two adjacent flaps apart, gently peeling the sclera from the retina. Repeat with the remaining cornea flaps, and remove the sclera from the retina (Figure 3B).
- Remove the lens from the retinal cup using the forceps. With microscissors, make 4 radial incisions from the edge of the retina towards the optic nerve, creating 4 equal petals (Figure 3B). Repeat steps 3.4-3.7 for the second eye.
4. Retinal flat-mount preparation
- Prepare grey mixed cellulose ester (MCE) membrane filter discs for mounting. If using large diameter MCE membrane filters, cut the disc into quadrants (roughly 1 cm across). Place the MCE disc onto the center of a larger white filter paper (Figure 3D).
NOTE: MCE filters are also available in 1 cm diameter discs. The MCE filter disc must be large enough to fit 1-2 retinas, but small enough to fit into the imaging chamber. As MCE membranes hold a static charge, minimize contact and handling of the MCE membrane. - Handling retinas using two size 3/0 paint brushes, flip one retinal cup onto a paintbrush with the retinal ganglion cell side down. Gently lift the retina out of the aCSF, making sure the water tension does not tear the retina (Figure 3C).
- While still holding the paintbrush with the retina, use a transfer pipette to place a droplet of aCSF in the center of the MCE filter paper (Figure 3C, right). Float the retina into the droplet of aCSF created by the surface tension. Use paint brushes to position the retina RGC side up within the droplet and to unfold the four petals.
- Once positioned, create a water bridge between the paintbrush and white filter paper to break the surface tension of the droplet.
NOTE: When aCSF is wicked away, the retina will adhere to the MCE filter paper (Figure 3D,E). Flat-mounted retinas can be handled by grasping the MCE disc with forceps. If the aCSF droplet quickly wicks away prior to forming a water bridge, this may indicate that the MCE membrane is not charged. Use a fresh MCE membrane, and minimize the time between enucleation and imaging by dissecting and flat-mounting retinas immediately before moving retinas into the imaging chamber.
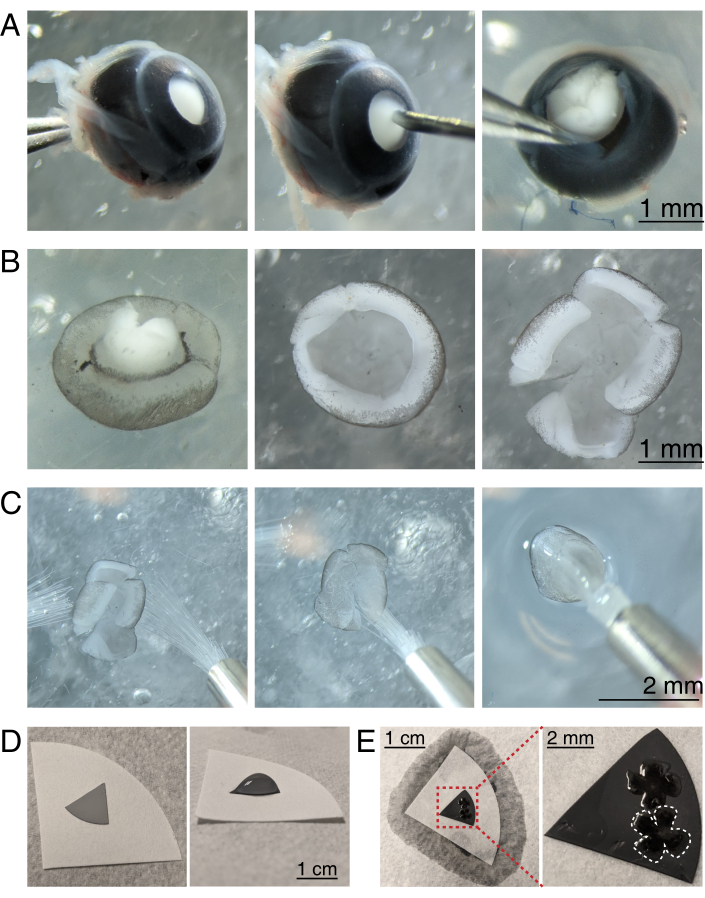
Figure 3: Retinal dissection and flat-mounting onto mixed cellulose ester filter membranes. (A) Left, an enucleated eye is stabilized by grasping the optic nerve with Dumont #5 forceps (left). A small hole is created in the center of the cornea using a 30 G needle (center). Micro-dissection scissors are used to cut the cornea into 4 equal flaps (right). Scale bar = 1 mm. (B) Dissected retina with the sclera peeled off using two Dumont #5 forceps (left) and with the lens removed (center). Retina with 4 equal cuts halfway into the retina (right). Scale bar = 1 mm. (C) Handling of retina with two fine paintbrushes (size 3/0, left). Retina flipped with retinal ganglion cells side down onto a paintbrush (center) and lifted out of aCSF, making sure the water tension does not tear the retina (right). Scale bar = 2 mm. (D) Grey MCE membrane disc placed onto a white filter paper (left). A droplet of aCSF on the MCE disc (right). Scale bar = 1 cm. (E) After floating the retinas into the droplet and positioning them, create a water bridge with the white filter paper to wick the aCSF, pulling the retina onto the charged MCE paper (left); scale bar = 1 cm. Zoomed image of the MCE membrane (red) shows 2 retinas mounted with retinal ganglion cells side up (right); scale bar = 2 mm. One retina is outlined in white. Abbreviations: MCE = mixed cellulose ester; aCSF = artificial cerebrospinal fluid. Please click here to view a larger version of this figure.
5. Time-lapse confocal imaging of live whole-mount retina preparations
- Assemble the live-imaging incubation chamber for an upright confocal microscope as seen in Figure 4.
NOTE: For inverted confocal systems, flat-mounts are placed RGC side down directly onto the glass bottom coverslip of the incubation chamber. Once the retinas make contact with the coverslip, they cannot be moved. - Fill the chamber with oxygenated aCSF, and turn on the pump and temperature controller (temperature 32-34 °C, flow rate 1 mL/min). Do not allow temperature to rise above 34 °C.
- To transfer the retinal flat-mount to the perfusion chamber, stop the pump, and remove the aCSF that is in the chamber. Place the MCE disc with the retinal flat-mount into the (empty) incubation chamber.
- Place a sample weight onto the flat-mount; pre-wet the weight to break surface tension. Refill the chamber with the warmed aCSF, and circulate aCSF at ~1 mL/min.
- Position the nosepiece with the 25x water dipping objective (numerical aperture 0.95) into the imaging chamber. Screen for labelled cells of interest using epifluorescent light (Figure 4C).
- Adjust the imaging volume to capture dendritic features of interest.
NOTE: This study captured the complete dendritic arbor at 1024 x 1024 pixels per frame, z-step 1 µm, and a frame rate of 2 min between each z-stack. Final image sizes are ~100 µm x 100 µm x 20 µm. - To adjust the laser power to an optimal setting, use a look-up table that identifies both oversaturated and undersaturated pixels. While scanning, adjust the laser power such that no pixels are oversaturated (i.e., at an intensity of 255 or above). Continue imaging as long as required, or until the there is a significant and detectable decline in fluorescent signal and increase in noise (typically 2-4 h).
NOTE: Reduced laser power is recommended as deconvolution algorithms work optimally when pixels are distributed over the full dynamic range. Pixel intensity should not exceed 254; empirical analyses of neurites revealed that pixel values below 170 are ideal for deconvolution. Fast scan speeds (400-600 Hz) with line averaging (2-3) are preferable to single, slower scans of the same total pixel dwell time. The area of prolonged imaging often photobleaches, but other explant sections remain viable. Multiple regions in a flat-mount can be imaged, each for 2-4 h, with a total incubation time of 6 hours. Imaging sessions beyond 6 h have not been systematically tested. Neurite degradation and blebbing are signs that the explant viability is declining. - After imaging, fix the retinal flat-mounts and membrane filter with cold 4% paraformaldehyde for 1-2 h at 4 °C. Amplify the fluorescent labels in the fixed retinas by immunohistochemistry for further analyses.
NOTE: Post-imaging fixation is not possible when using an inverted confocal; retinas are not removable without destroying the tissue.

Figure 4: Live-cell imaging incubation chamber setup. (A) Live-imaging incubation apparatus showing heating, solution, and imaging components. Dual heater includes a heated incubation chamber platform (back circle with blue connector electrodes) and an in-line solution heater. (B) Schematic diagram of 4A. Retinal flat-mount (red) on MCE membrane (grey) is placed in the incubation chamber. The in-line solution heater is connected to a solution pump (not pictured in 4A). Imaging chamber dimensions allow a good working area for the dipping objective nose. (C) 25x view a retinal explant injected with 0.23-0.45 µL of 4 × 1012 GC/mL of AAV dilution; scale bar = 100 µm. Dense labeling of cells often surrounds the optic nerve head (dashed line, bottom right); scale bar = 50 µm. Abbreviations: MCE = mixed cellulose ester; GC = genome copies; mCherry = monomeric Cherry; eYFP = enhanced yellow fluorescent protein. Please click here to view a larger version of this figure.
6. Image deconvolution and post-processing in ImageJ
- Import the image series, split the series by time, and color if it is a multi-color image. Ensure that all time points for one color are contained in the same folder.
NOTE: Import Bio-formats if using ImageJ (Bio-formats is automatically included in FIJI). - Create a theoretical point-spread function (PSF) using the ImageJ plugin, Diffraction PSF 3D.
NOTE: Each imaging channel requires its own PSF as fluorescent protein emission wavelength impacts the PSF. - Use Plugins | Macros | Run to perform batch Parallel Iterative Deconvolution for all timepoints using the provided macro (Supplementary File 1).
NOTE: This macro runs 25 iterations of the Wiener Filter Preconditioned Landweber (WPL) iterative algorithm. Each color channel must be deconvolved separately. - Merge the color channels, and compile all time points into a Hyperstack (Image | Hyperstacks | Stacks to Hyperstack). Correct the 3D drift using Plugins | Registration | Correct 3D Drift.
- Return the image to a regular stack (Image | Hyperstacks | Hyperstack to Stack), and split the time points (Image | Stacks | Tools | Stack Splitter). Use batch processing to create maximum projection for all time points (Process | Batch | Macro | run("Z Project…", "projection=[Max Intensity]"); ).
- Import the time-lapse image sequence (File | Import | Image Sequence). Use conventional ImageJ tools for desired analysis of deconvolved and post-processed two-dimensional (2D) video.
NOTE: Four-dimensional (3D + time) deconvolved and drift-corrected videos can be viewed as a hyperstack. Omit the previous step to maintain 3D time points.
Using the above protocol, a high-resolution 3D video of developing starburst cell dendrites was acquired, deconvolved, and corrected for 3D drift. Z-plane maximum projections were produced to make 2D videos for analysis (Supplementary Video 1, Figure 5A). 3D deconvolution of each time point increased the resolution of fine filopodia projections (Figure 5B,C). Fine filopodia protrusions are a feature of developing retinal dendrites36 and should be visible during imaging. AAV transfection will produce variability in cell labeling density and fluorescence intensity. Thus, screen the labelled tissue to select cells with high fluorescent signals for imaging and producing high-quality videos. Increasing the laser power to detect dimly labelled cells is not recommended, as it can lead to rapid photobleaching and introduce high noise compared to the signal. Sufficiently bright cells are visible within 5 dpi. By 12 dpi and beyond, prolonged AAV infection periods do not necessarily lead to increased fluorescence of labeled cells (Figure 5D).
Excessive fluorescence and neurite crowding from dense cell labelling affects image quality and can hinder single-cell reconstructions. Optimization of the AAV dilution and volume is required to achieve the desired degree of isolated and fluorescently labeled arbors that are separable from the rest of the target cell population. Here, 9.2 × 108-1.8 × 109 viral GCs were injected per eye to target starburst amacrine cells (0.23-0.45 µL of AAV with a 4 × 1012 GC/mL titer). The absence of a fluorescent signal might reflect an inefficient injection technique. While injecting, leakage of the blue AAV solution from the injection site indicates that the virus is not delivered to the retina, or that the injection volume or pressure is too high. Excess injection volume or pressure can cause retinal detachment and/or loss of ocular pressure, damaging the retinal cells and reducing cell-labeling efficiency. Both issues can be resolved with practice and optimization of the injection technique. Other potential reasons for the lack of signal include degradation of the AAVs from improper storage or insufficient Cre expression because the Cre transgene is not active during the period of AAV transfection.
Once videos are deconvolved, dynamic analyses, such as calculating rates of branch addition, retraction, or stabilization, can be performed. Additionally, conventional static analysis can be performed on each timeframe, including neuron reconstruction and morphometric quantifications such as total branch length, branch angles, branch order, or number of terminal branch points. The result of this protocol is a time series of high-resolution 3D image stacks. Most 3D neurite dynamic videos are manually quantified as a maximum projection using an open-source image visualization software such as ImageJ37. Another open-source tool, Vaa3D38, has been developed specifically for visualization of large volumes. If videos are to be analyzed in 3D, use of a 3D + time visualizer such as Vaa3D is recommended. ImageJ and Vaa3D allow direct access to the pixels or voxels in the image, but often neuron reconstructions are created by converting pixel data into skeletonized tree reconstructions. Automatic or semi-automatic neuron reconstructions for single time points can be performed with open-source39,40,41 and proprietary software. To analyze time-lapse reconstructions, each point on the reconstruction must be linked to the previous and the subsequent time points. Aligning complex dendritic morphologies over time, which can be encoded by upwards of 500 data points, remains a challenging problem. Many tools are available, with different strengths and limitations, and they must be selected to suit the analyses needed for the study.
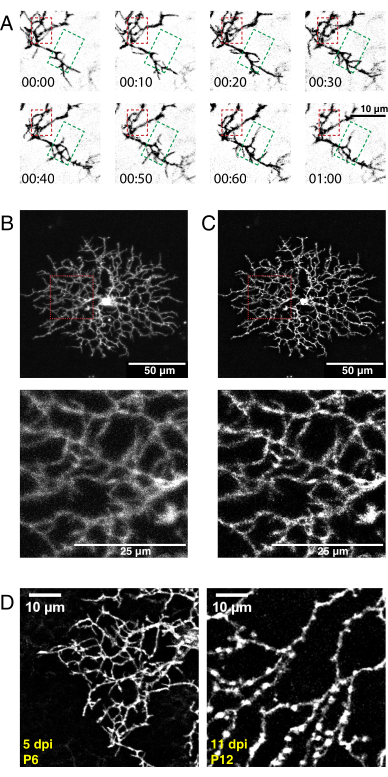
Figure 5: Representative results after neonatal intraocular injection, time-lapse imaging, and deconvolution. (A) Maximum projections of deconvolved time-lapse imaging series (h:min) of P6 starburst amacrine cell 5 days postAAV injection. Fine filopodia dynamics can now be analyzed using conventional ImageJ tools. An area of dendritic refinement (red box) and an area of dendritic outgrowth (green box) are highlighted. (B, C) Single P6 starburst amacrine cell labelled with membrane-tagged eYFP 5 days postAAV injection (dpi) shows bright single-cell labelling. Scale bars for upper panels = 50 µm; scale bars for lower panels = 25 µm. Before (A) and after (B) deconvolution with ImageJ. Insets (red) show deblurring as a result of successful deconvolution. (D) P6 starburst amacrine cell 5dpi (left) compared to P12 starburst cell 11 dpi shows similar levels of fluorescent protein expression. Increasing AAV incubation times do not increase fluorescent signal. The fluorescent signals do not decline with longer AAV infection periods. Identical acquisition and postprocessing parameters were applied to both images. Increased branch thickness and varicosities observed at P12 are normal features of maturing starburst cells and not a labelling or imaging artifact. Scale bars = 10 µm. Abbreviations: AAV = adeno-associated virus; eYFP = enhanced yellow fluorescent protein; dpi = days postinfection. Please click here to view a larger version of this figure.
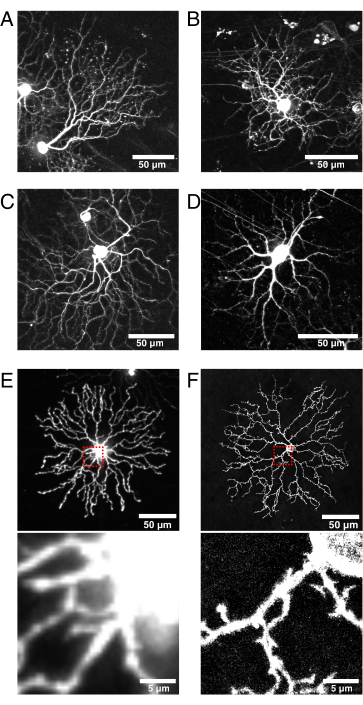
Figure 6: Considerations for Cre line selection and fluorescent protein localization.
(A–D) Live P11 retinal ganglion cells imaged at 9 dpi of the BBV injected into Vglut2-iRES-Cre knock-in mice, in which Cre is selectively expressed in the RGC population. (A) JAM-B RGC identified by its characteristic asymmetric morphology24. (B–D) Distinct RGC subtypes that differ in their dendrite stratification patterns. (E) Image of live P11 starburst amacrine cell biolistically transfected with cytosolic mCherry and captured with spinning disk confocal at 512 x 512 pixels. The cytosolic XFP leads to high fluorescence signal in the soma relative to the fine dendritic protrusions. Out-of-focus light from the soma causes high background fluorescence when imaging small proximal projections (inset in red box above is shown below). (F) Image of live P11 starburst amacrine cell transfected with membrane-targeted eYFP and captured by confocal laser scanning at 1024 x 1024 pixels. Note the fluorescent membrane ring around the soma, the improved signal-to-noise ratio, and quality of the fine projections proximal to the soma that are produced by membrane-targeted fluorescent proteins (inset). Scale bars = 50 µm and for lower panels of E and F = 5 µm. Abbreviations: IRES = internal ribosome entry site; BBV = Brainbow adeno-associated virus; Vglut2 = vesicular glutamate transporter 2; XFP = any fluorescent protein; eYFP = enhanced yellow fluorescent protein; mCherry = monomeric Cherry; RGC = retinal ganglion cells; JAM-B = junction adhesion molecule B. Please click here to view a larger version of this figure.
Supplementary Video 1: Time-lapse video of P6 developing starburst amacrine dendrites, 5 days postinjection. Video time-step is 2 min. Deconvolution and 3D drift correction were applied using ImageJ. Top yellow boxes highlight areas of outgrowth, and the lower box outlines a region of transient filopodia outgrowth, self-contact, and retraction. Please click here to download this Video.
Supplemental File 1: ImageJ macro for batch Parallel Iterative Deconvolution. The macro runs 25 iterations of the Wiener Filter Preconditioned Landweber (WPL) iterative algorithm. Please click here to download this File.
