Traditional worm lysis methods have various disadvantages. For example, probe-based sonication and bead-beater methods produce excessive heat by allowing contact of the metal tip or beads directly with the samples, resulting in variable protein recoveries and protein denaturization. Liquid nitrogen grinding followed by sonication in lysis buffer, can be time-consuming and requires a large number of worms. Due to the limitations of traditional worm lysis methods, previous MS workflows, such as the labeling methods iTRAQ or label-free methods that have been historically used in the C. elegans model system to gain quantitative information about the insolublome, require large input of starting material (at least 40,000 worms). Laborious worm culture work is required to obtain these numbers of worms. Moreover, the labelling methods require expensive isobaric chemical labels. Label-free quantification methods are cost-effective and have easier and more straightforward sample preparation and labeling methods, but require significantly large numbers of worms to achieve sufficient MS analysis coverage.
The sonicator that we used greatly increases the efficiency and reproducibility of worm lysis by lysing multiple worm samples simultaneously in a temperature-controlled water bath sonicator without cross-contamination14, thus significantly reducing the amount of starting worm material required. Combining the highly efficient sonication method and the quantitative DIA label-free MS approach, we were able to robustly quantify the insolublome of aged and young worms using ~3,000 worms. Here we tested and validated the efficiency of the protocol and compared the insolublome of aged and young worms from a wild-type worm strain, N2-Bristol C. elegans. We applied this protocol to extract and isolate the insolublome from ~3,000 aged and young N2 C. elegans (two biological replicates for each condition), followed by MS analysis with a quadrupole time-of-flight mass spectrometer or other MS systems using a combination of data-dependent acquisition (DDA) and data-independent acquisitions (DIA/SWATH) for protein identification and quantification. The insoluble proteins were first analyzed on a Bis-Tris 4-12% gradient gel to determine the amount of protein in each insolublome sample. As demonstrated in Figure 2, the insolublome sample from N2 aged worms (lanes 2 and 3, biological replicate experiments) has significantly more protein than samples from the N2 young worms (lanes 1 and 4, biological replicate experiments).
After in-gel digestion, the protein profiles of the insolublome were analyzed by HPLC-MS. Using this workflow, we can generally identify 1000–1500 proteins and quantify 500–1,000 proteins from the SDS-insoluble fraction with high reproducibility (unpublished data). Here we were able to quantify 989 proteins from the insolublome of N2-Bristol C. elegans by analyzing the DIA data and removing redundancy: 768 proteins were significantly enriched and 27 proteins were significantly decreased in the insolublome of aged N2 worm (Day 10) compared with young (day 2) using a fold-change of at least 1.5 and a Q value of less than 0.01 (Figure 3A). As seen on the histogram plot (Figure 3B), the fold-change of significantly altered proteins shows a normal distribution. Aged worms were demonstrated to be significantly enriched for the insolublome: The largest change observed showed the relative protein abundance in the insolublome to be 592 times higher in the old versus young worms; and for 32 proteins the relative protein abundance in the insolublome was >250 times higher in the old versus young worms indicating dramatic insolublome changes with age.
After extracting the list of insoluble proteins that are significantly increased in the aged worms and identified by the wormbase (WS271), KEGG pathway and Gene Ontology (GO) analysis were performed to determine the pathways that are enriched in the aged insolublome to gain biological insights into how these relate to aging. The KEGG pathway analysis of proteins identified in this study shows enrichment of several pathways involving ribosomes, mitochondria, proteasome and spliceosome (Figure 4A). The gene ontology analysis shows that the insolublome from aged worms comprises many proteins in particular categories including mitochondrial, developmental, determinants of adult lifespan, and ribosomal proteins (Figure 4B and Supplementary Table 1A). We then compared the list of proteins identified in this study with previously published work from David et al.2 and Mark et al.11 as demonstrated in Venn diagrams (Figure 5A,5B). The comparison showed significant overlap of identified proteins 394/721 and 444/721 with David et al (Figure 5A) and Mark et al. (Figure 5B) study, respectively. The biological pathways revealed by the KEGG analysis of insolublome from this study have also been identified in the past thus validating our methodology (Supplementary Table 1B). Identification of these pathways and proteins suggests that they may serve as candidates for further biological investigation in regards to their function in the context of aging.
In summary, the use of the efficient sonication method enables the lysis of multiple worm samples at the same time in an environment with well-controlled temperatures and reduced cross-contamination to achieve high protein coverage with significantly less starting worm material. Combining the efficient sonication method with a DIA label-free protein quantification workflow has provided reliable and reproducible results for the quantification of insoluble worm proteins.
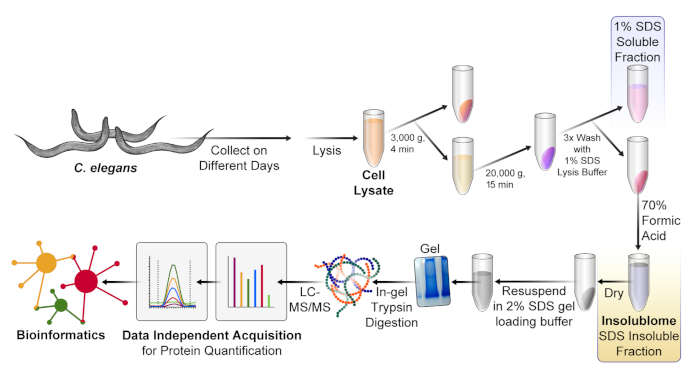
Figure 1. Experimental workflow of the protocol. C. elegans were cultured and collected on different days. After worm lysis with a sonicator, the 1% SDS-insoluble protein fraction (insolublome) was extracted and isolated from the lysate. The insolublome was then digested via in-gel trypsin digestion and quantified via DIA mass spectrometry, followed by bioinformatic analysis. Please click here to view a larger version of this figure.
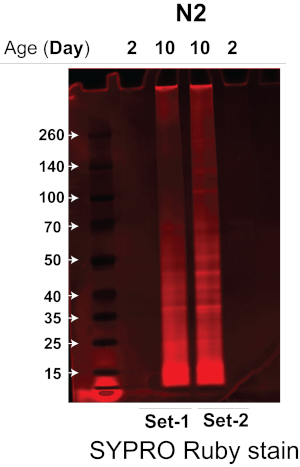
Figure 2. SDS-PAGE gel of the insolublome isolated young versus old worms of the N2-Bristol strain. The insolublomes of young versus old worms of the N2-Bristol strain were analyzed by SDS-PAGE to determine the amount of protein present. The SDS-PAGE gel was stained with a fluorescent protein stain to visualize protein bands. Lanes 1 and 4: Insolublome from two biological replicate experiments of young N2 worms (Day 2). Lanes 2 and 3: Insolublome from two biological replicate experiments of aged N2 worms (Day 10). Please click here to view a larger version of this figure.
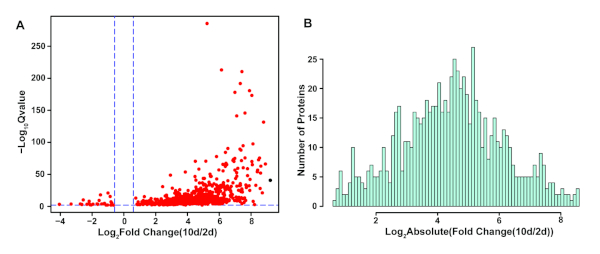
Figure 3. Protein candidates identified as showing significant alteration in the aged versus young insolublome and their fold-change distributions. (A) Volcano plot for quantification of the insolublome of aged versus young N2-Bristol worms. Candidates with an absolute fold change >=1.5 and Q value <0.01 are shown as red dots. (B) Histogram plot for fold-change distribution of significantly enriched SDS-insoluble proteins in aged versus young worm samples. Please click here to view a larger version of this figure.
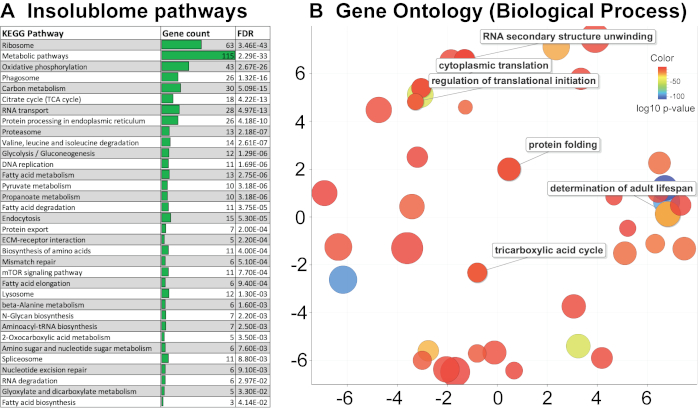
Figure 4. KEGG pathway and Gene Ontology (GO) analysis. (A) KEGG pathway analysis of the day-10 insolublome arranged according to p-value with highly significant pathway shown at the top. (B) Gene Ontology analysis shows that insolublome of aged worms is enriched for many proteins in particular categories including mitochondrial, developmental, determinants of adult lifespan, and ribosomal proteins. The scatterplot view visualizes the GO terms in a “semantic space” where the more similar terms are positioned closer together. The color of the bubble reflects the p-value obtained in the STRING analysis, while its size reflects the generality of the GO term in the UniProt-GOA database. Please click here to view a larger version of this figure.
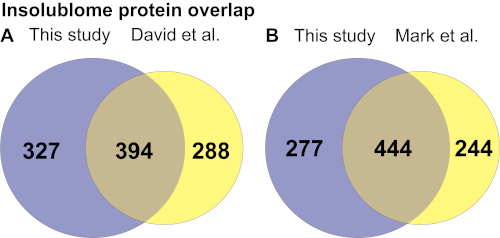
Figure 5. Insolublome protein overlap identified in day-10 insolublome comparing this study with (A) David et al.2 and (B) Mark et al.11 studies. Please click here to view a larger version of this figure.
Supplementary Table 1 (related to Figure 4 and Figure 5). (A) Gene ontology (biological process) analyzed with STRING database. (B) Detailed list of proteins and KEGG pathways identified in this study with color codes depicting their overlap with the published work. Please click here to download this file.
