Using this protocol, we treated three mice with a 10 mg/kg dose of Cy5.5-CTP through a retro-orbital injection (Figure 1A). After allowing the peptide to circulate for 15 min, the mice were euthanized using a CO2 chamber, the chest opened through a median sternotomy incision, the right atrium was nicked, and the mice perfusion fixed using 3 mL of 10% phosphate-buffered formalin (Figure 1B,C). After fixation, the heart, lungs, liver, kidney, spleen, and brain were dissected out and arranged in a 12 well plate for ex vivo optical fluorescent imaging (Figure 1D and Figure 2A). Three control mice with no injections were also perfused, dissected, and imaged (Figure 1B−D and Figure 2B). The fluorescence data acquired for each set of organs can be compared due to the counts being converted to radiant efficiency, the relative fluorescence unit of the imaging software. This data can be quantified to yield both qualitative images and quantitative data for each organ in question (Figure 2C) across different time points for biodistribution studies. The autofluorescence of different organs in response to different excitation wavelengths is demonstrated in Figure 3A−E. Shorter excitation wavelengths, such as enhanced green fluorescent protein (EGFP), are associated with higher autofluorescence, especially in liver and brain, than far-red (Cy5.5) or near infrared (Cy7) excitation wavelengths (Figure 3). The imaged organs were fixed in 10% phosphate buffered formalin at RT for a minimum of 48 h, followed by a transfer to tissue processing and embedding cassettes (Figure 1E). The organs were processed and paraffinized in a tissue processing machine, positioned in molds filled with paraffin, frozen on a -20 °C stage, and stored overnight at -20 °C (Figure 1F). The samples were sectioned (Figure 1G) and treated with solution exchanges to rehydrate the tissue (Figure 1H). The prepared slides were then mounted with DAPI fluorescent mounting medium (Figure 1I). Once dry, usually overnight, the slides were imaged using fluorescent microscopy. Representative images of each organ from a mouse are shown in Figure 4A. The images from different organs were acquired using identical settings to allow for comparison across mice, and to allow for quantification (Figure 4B).
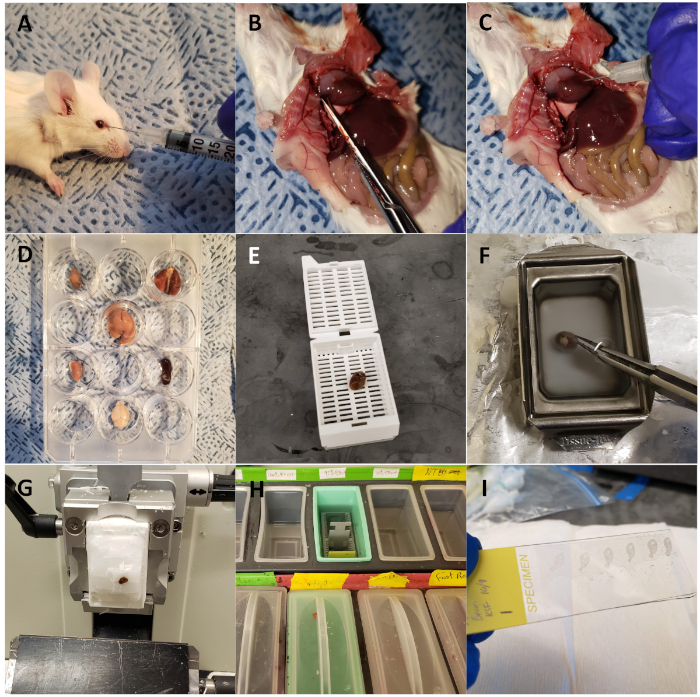
Figure 1: Harvesting mouse organs for ex vivo optical fluorescent imaging and sectioning. (A) Wild type mouse injected retro-orbitally with CTP-Cy5.5. (B) Mouse dissected with chest opened, right atrium snipped, for perfusion fixation. (C) Heart injected with 3 mL of 10% buffered formalin phosphate for perfusion fixation of the animal. (D) Organs of interest harvested and arranged in a 12 well plate for fluorescent optical imaging. (E) Heart loaded into a cassette and processed using a tissue processor. (F) Heart embedded in paraffin. (G) Heart sectioned on a microtome. (H) Slides deparaffinized through a series of solution exchanges. (I) Sections mounted with coverslips using DAPI. Please click here to view a larger version of this figure.
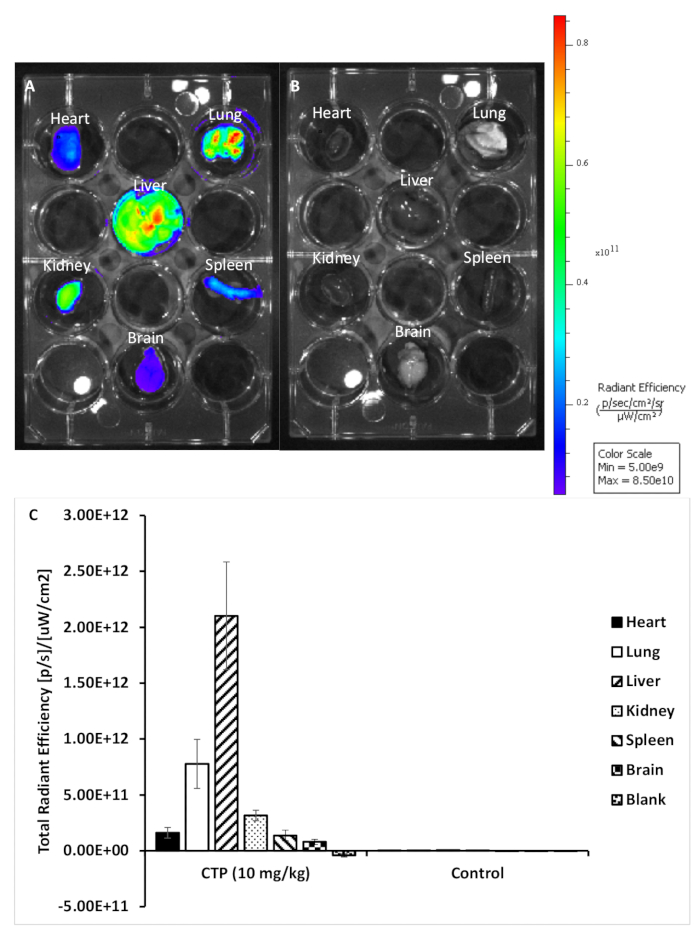
Figure 2: Representative images from treated and control organs. (A) Organs from a mouse treated with 10 mg/kg CTP-Cy5.5. (B) Organs from an untreated control mouse. (C) Quantification of fluorescence for each set of organs. Please click here to view a larger version of this figure.
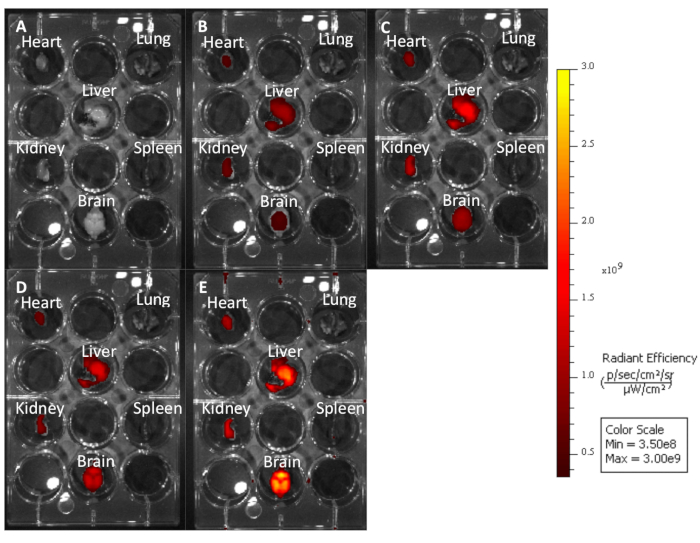
Figure 3: Untreated mouse organs imaged at different excitation emission wavelengths to demonstrate how longer wavelengths produce less autofluorescence. (A) Cy7: 740−790. (B) Cy5.5: 660−710. (C) Cy5: 620−670. (D) Cy3: 520−570. (E) EGFP: 480−520. Please click here to view a larger version of this figure.
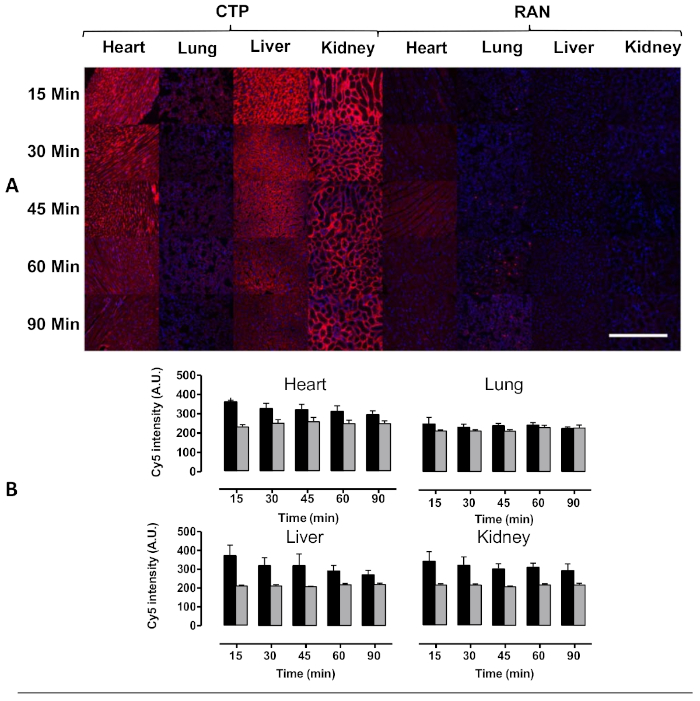
Figure 4: Transduction of heart, lung, liver, and kidney after intravenous injection in mice. Wild type mice were injected with 10 mg/kg of Cy5.5-CTP, or a random peptide (RAN-Cy5.5), and euthanized at the indicated time points. (A) Peak transduction of heart tissue was seen at 15 min with steady decrease in fluorescence over time. Some capillary uptake was noted in the lungs with robust transduction in the liver as well at the kidney glomerular capillaries, the latter implying a renal mechanism of excretion. (B) Quantification of fluorescent intensity shows significantly increased heart uptake of CTP-Cy5.5 over RAN-Cy5.5. Scale bar = 500 µm. This figure has been modified from Zahid et al.8. Please click here to view a larger version of this figure.
