Based on the different birthdates of neuronal subtypes in the cerebellum, cultures from E12−E18 mouse embryos yielded different cell types. Large projection neurons, such as CN neurons (E9−E12) and PCs (E10−E13), emerged early during cerebellar development. In mice, granule and Golgi cells arose between ~E13–E18 and underwent terminal divisions up to postnatal week 4.
Replacing old medium I with fresh culture medium II at days in vitro (DIV) 7 will eventually prevent glial cell proliferation. The interneurons of the molecular layer, such as basket and stellate cells, differentiate after birth. Therefore, most of the cells in the culture medium at E18 were expected to be a combination of PCs, granule cells, CN, and some Golgi cells. Calbindin 1 (CALB1), a specific marker of PCs, was used to track the morphological changes during the 21-day time course. On DIV 0, the cell bodies of PCs were detectable (e.g., 10−11 h of incubation), but the outgrowth of neurite had not yet started (Figure 2A). By DIV 3, the axonal extension showed progress, while dendritic processes had just started. This status remained almost unchanged until the second week in vitro (WIV) (Figure 2B–D). On DIV 10, a few sparse branches and primary dendrites with spines started growing (Figure 2E). Sprouting of the new primary dendrites occurred continuously after DIV 14. Therefore, after DIV 14, the number of secondary and tertiary dendrites increased and developed wide branches at DIV 21 (Figure 2F,G).
It is important to know that the timing of the morphological changes in vitro may not follow the same pattern as in vivo. Following these results, in another experiment the dissociated cerebellar primordia from E12, E13, E14, and E15 were cultured for 3 weeks. The cultured PCs from E12 and E13 did not develop any dendritic outgrowth on DIV 18 except axonal extensions (Figure 3A,B). However, after the same period of time, the dissociated cerebellar primordium cultures from E14 and E15 showed dendritic outgrowth and arborization (Figure 3C,D). This growth rhythm variation among neural cells in vitro sheds light on a big change that happened in their natural environment between the early and late stage of cerebellar development, which needs to be applied in vitro for medium optimization.
Along with PCs, there are other neuronal cell types in the cerebellum that develop during dissociated primary cerebellar cultures that can be detected using specific neuronal markers. Double immunofluorescence labeling for CALB1 and a calcium-binding albumin protein, parvalbumin (PVALB), showed that CALB1 expression was exclusively restricted to PCs in primary cerebellar cultures, while PVALB was expressed in CALB1+ neurons (i.e., PCs) and PVALB+/CALB1– neurons, which are molecular layer interneurons (basket/stellate cells) (Figure 4A–C). The alpha subunit (Nav1.6) of the voltage-gated sodium channel (SCN) is a marker for granule cells and PCs in the cerebellum20. Double labeling with anti-SCN and anti-CALB1 shows the colocalization of granule cell bodies and PCs in culture medium on DIV 21 (Figure 4D–F). For other specific cerebellar cell types from dissociated primary cerebellar cultures, please see Marzban and Hawkes19.
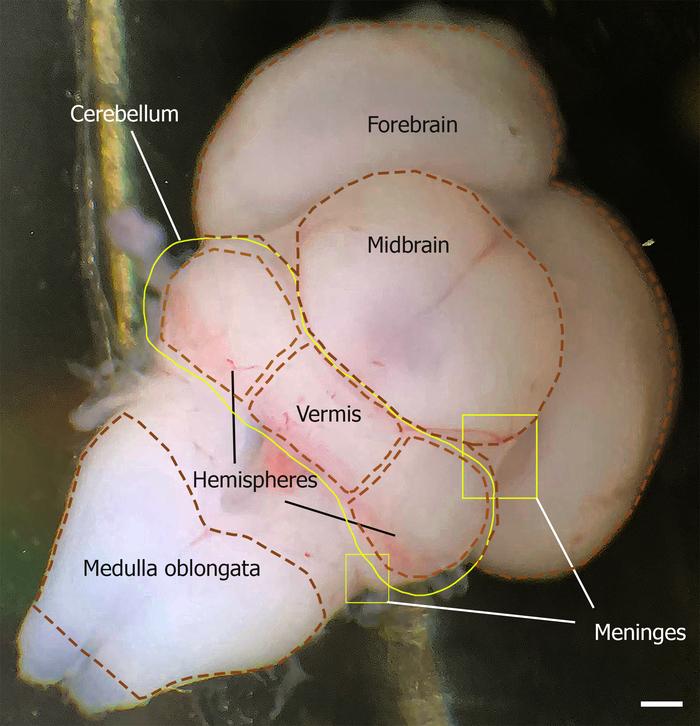
Figure 1: Dorsal aspect of the mouse brain outlining the cerebellum at E18. The meninges are indicated by yellow squares. The location of the cerebellum is outlined by the yellow line, which is limited rostrally by the midbrain and caudally by the medulla oblongata (outlined by brown dashed lines). The cerebellum vermis and hemisphere are shown by brown dashed lines. Please click here to view a larger version of this figure.
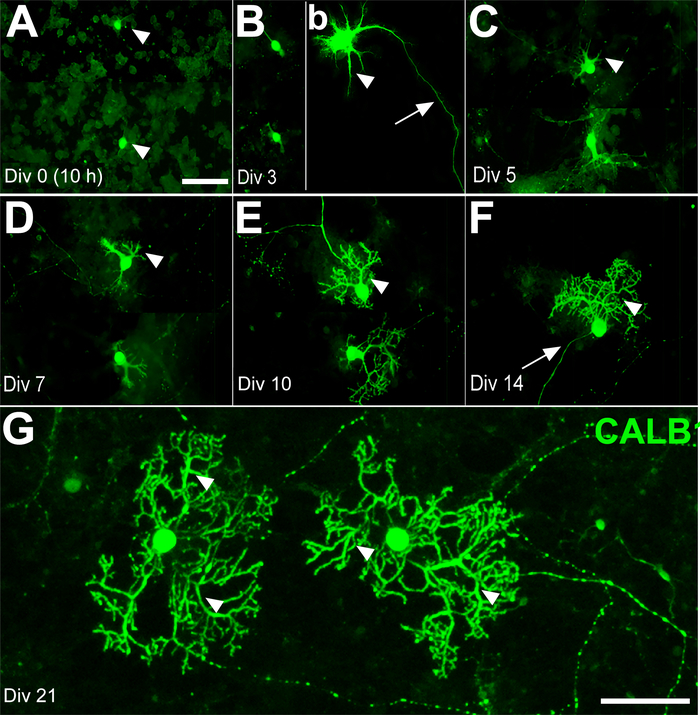
Figure 2: Development of Purkinje cells (PCs) derived from the mouse cerebellum at E18 in primary culture for days in vitro (DIV) 0–21. (A) PC somata (arrowheads) were labeled by immunofluorescence using anti-calbindin 1 (CALB1) at DIV 0 (after 10 h). (B) The first axonal extension (arrow) and early dendritic outgrowth (arrowhead) appear on DIV 3. The PCs’ dendritic outgrowth and development (arrowhead) continue on DIV 5 (C) and DIV 7 (D). The PCs’ dendritic branches are clearly distinguishable on DIV 10 (E) and DIV 14 (F) (arrowheads) and elaborate dendritic trees are detectable at DIV 21 (arrowhead) (G). Scale bars: A = 100 μm (applies to panels B, C, D, E and F); G = 50 μm. Please click here to view a larger version of this figure.
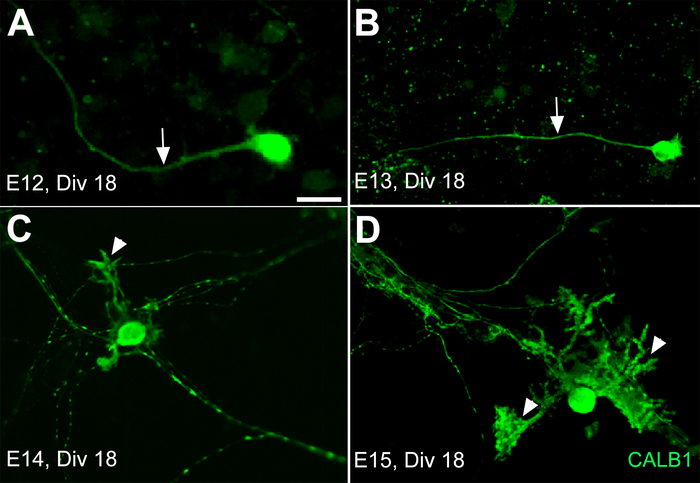
Figure 3: Development of Purkinje cells (PCs) derived from the mouse cerebellar primordium at E12, E13, E14, and E15 after 18 days in primary culture (DIV 18). The axons are the only extensions (arrow) from the PC somata in primary culture of the E12 and E13 cerebella primordium after 18 days in vitro (DIV 18) (A,B). The PCs’ dendrite outgrowth and extension develop by DIV 18 (arrowhead) only from E14 (C) and E15 (D) cerebellar primary cultures. Scale bar = 20 μm (applies to panels A−D). Please click here to view a larger version of this figure.
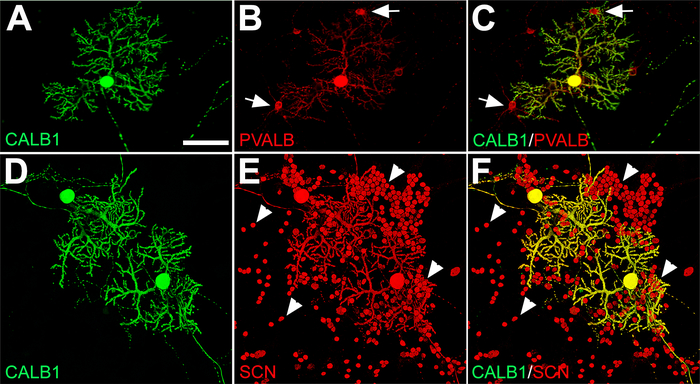
Figure 4: Immunofluorescence labeling of PCs, GABAergic interneurons (basket and stellate cells), and granule cells from E18 mouse cerebellar primary culture at DIV 21. (A−C) Double labeling with anti-CALB1 (green) and anti-PVALB (red) shows PCs and a few interneurons (arrow) in contact with PC dendritic arbors. (D−F) Voltage-gated sodium channels (SCNs) in PC bodies with elaborated dendrites and numerous small granule cell bodies (arrowhead) are labeled with anti-SCN (red) and double-labeled with anti-CALB1 (green). Abbreviations: Purkinje cells = PCs; CALB1 = calbindin 1; PVALB = parvalbumin; SCN = voltage-gated sodium channel. Scale bar = 100 μm (applies to A−F). Please click here to view a larger version of this figure.
| Name | Basic medium | Putrescine | Sodium selenite | L-glutamine | Gentamicin | T3 | N3 | BSA | Ara-C | FBS |
| Culture medium I | DMEM/F12 | 100 µM | 30 nM | 3.9 mM | 3.5 µg/mL | 0.5 ng/mL | Progesterone 4 µM, Insulin 20 μg/mL, Transferrin 20 mg/mL | – | – | – |
| Seeding medium | DMEM/F12 | 100 µM | 30 nM | 3.9 mM | 3.5 µg/mL | – | – | – | – | 10% with culture medium I |
| Culture medium II | Culture medium I | 100 μg/mL | 4 μM | – | ||||||
Table 1: Media compositions.
