Antibody-Free Assay for RNA Methyltransferase Activity Analysis
Summary
Here, an antibody-free in vitro assay for direct analysis of methyltransferase activity on synthetic or in vitro transcribed RNA is described.
Abstract
There are more than 100 chemically distinct modifications of RNA, two thirds of which consist of methylations. Interest in RNA modifications, and especially methylations, has re-emerged due to the important roles played by the enzymes that write and erase them in biological processes relevant to disease and cancer. Here, a sensitive in vitro assay for accurate analysis of RNA methylation writer activity on synthetic or in vitro transcribed RNAs is provided. This assay uses a tritiated form of S-adenosyl-methionine, resulting in direct labeling of methylated RNA with tritium. The low energy of tritium radiation makes the method safe, and pre-existing methods of tritium signal amplification, make it possible to quantify and to visualize the methylated RNA without the use of antibodies, which are commonly prone to artifacts. While this method is written for RNA methylation, few tweaks make it applicable to the study of other RNA modifications that can be radioactively labeled, such as RNA acetylation with 14C acetyl coenzyme A. Overall, this assay allows to quickly assess RNA methylation conditions, inhibition with small molecule inhibitors, or the effect of RNA or enzyme mutants, and provides a powerful tool to validate and expand results obtained in cells.
Introduction
DNA, RNA and proteins are subject to modifications that tightly regulate gene expression1. Among these modifications, methylations occur on all three biopolymers. DNA and protein methylations have been very well studied during the last three decades. In contrast, interest in RNA methylation has been only recently reignited in light of the important roles that proteins that write, erase or bind RNA methylations play in development and disease2. In addition to better known functions in the abundant ribosomal and transfer RNAs, RNA methylation pathways regulate specific messenger RNA stability3,4, splicing5 and translation6,7, miRNA processing8,9 and transcriptional pausing and release10,11.
Here, a simple and robust method for in vitro verification of RNA methyltransferase activity in the setting of a molecular biology lab is reported (summarized in Figure 1). Many studies assess the activity of an RNA methyltransferase through dot-blot with an antibody against the RNA modification of interest. However, dot-blot does not verify the integrity of the RNA upon incubation with the RNA methyltransferase. This is important because even minor contaminations of recombinant proteins with nucleases can lead to partial RNA degradation and confounding results. Moreover, even highly specific RNA modification antibodies can recognize unmodified RNAs with specific sequences or structures. The in vitro RNA methyltransferase assay reported here takes advantage of the fact that the S-adenosyl methionine can be tritiated on the methyl group donor (Figure 1), allowing for the methylated RNA to be accurately detected without the use of antibodies. Instructions are provided for in vitro transcription and purification of a transcript of interest and testing of the methylation of said transcript by the enzyme of interest. This method is flexible and robust, and can be adjusted according to the needs of any given project. For example, in vitro transcribed and purified RNAs, chemically synthesized RNAs, but also cellular RNAs can be used. This assay provides quantitative information in the form of scintillation counts, as well as qualitative information by showing where exactly the methylated RNA runs on a gel. This can provide unique insight into the function of an RNA methyltransferase, particularly when using cellular RNAs as a substrate, as it provides a method to directly observe the size of the RNA or RNAs that are targeted for methylation.
Protocol
1. In vitro transcription and gel purification of the target RNA
- Clone the sequence of interest into plasmids containing T7 and/or SP6 promoters using established molecular cloning techniques12 or kits.
- Linearizing the DNA template for in vitro transcription
- Amplify the sequence of interest by PCR of the plasmid with primers designed to include the T7 promoter region through the insert, +20-30 bp upstream and downstream as previously described13.
- Alternatively, digest 5 µg of the plasmid using a restriction enzyme (RE) cut site available downstream the insert according to the instructions provided by the supplier of the RE.
NOTE: Remember to double-check that the insert is not cut by the selected RE. Selection of the RE used in this step can dramatically affect the yield of the transcript. Preferentially, linearize with a blunt or 5'-overhanging end producing RE to avoid template switching of the T7 RNA polymerase on the opposite DNA strand14. Try different RE if initial yields are insufficient.
- Purify the resulting DNA using the kit for DNA agarose gel extraction and clean-up. Elute the DNA with 30 µL of water.
- Mix well by vortexing, pipet 1.5 µL and measure DNA concentration with a microvolume spectrophotometer.
- Use 250 ng of DNA to check the quality and size of the purified DNA on a 1% agarose gel electrophoresis in 1x TBE buffer migrated for 1 h at 4 V/cm.
- Thaw the frozen reagents of the in vitro transcription kit. Place the T7 RNA Polymerase Mix on ice, and the other reagents on a nutator at room temperature. Immediately after complete thawing, quick spin the rNTP tubes for 5 s to collect solution at the bottom of the tubes and place on ice. Keep the 10x Reaction Buffer at room temperature to avoid precipitation.
- Pipet into a 1.5 mL tube the following components of the 20 µL transcription reaction at room temperature in the indicated order: 6 µL of water, 2 µL (0.1 – 1 µg) of linear DNA template, 2 µL of 75 mM ATP solution, 2 µL of 75 mM GTP solution, 2 µL of 75 mM CTP solution, 2 µL of 75 mM UTP solution, 2 µL of 10x Reaction Buffer and 2 µL of T7 RNA Polymerase Mix.
NOTE: For DNA template generated by PCR, use 100 - 200 ng DNA; for DNA template generated by restriction enzyme digest of a plasmid, use ~1 µg. Active recombinant His6-tagged T7 RNA Polymerase can be purified in E. coli15. - Mix the tube thoroughly. Quick spin for 5 s to collect reaction solution at the bottom of the tube, then incubate at 37 °C for 2-4 h.
NOTE: Every transcript will behave differently depending on the DNA template, its length, its sequence or structure. Optimize in vitro transcription conditions by testing different incubation times up to 6 h; different concentrations of DNA template, T7 RNA Polymerase, or NTP; or addition of supplementary MgCl2 to what is present in the T7 RNA Polymerase Mix. - At the end of the incubation period, add 1 µL of DNase I per 20 µL reaction and incubate at 37 °C for 15 min.
NOTE: The transcription reaction can be temporarily kept on ice until the polyacrylamide gel is ready. - Proceed with purification by polyacrylamide gel.
NOTE: Since RNA is sensitive to pH and RNase contamination, use RNA dedicated equipment and RNase-free reagents. - Determine the percentage of polyacrylamide for the gel depending on the size of the transcript of interest: 3.5% for 100-2000 nt (XC: 460 nt ; BB: 100 nt), 5% for 80-500 nt (XC: 260 nt ; BB: 65 nt), 8% for 60-400 nt (XC: 160 nt ; BB: 45 nt), 12% for 40-200 nt (XC: 70 nt ; BB: 20 nt), 15% for 25-150 nt (XC: 60 nt ; BB: 15 nt), and 20% for 6-100 nt (XC: 45 nt ; BB: 12 nt).
- Prepare the urea denaturing polyacrylamide gel by combining the following reagents in a 50 mL conical tube: 9.6 g of molecular grade urea, 2 mL of 10x TBE, x mL of 40% acrylamide:bis-acrylamide mix (29:1) and water up to the 20 mL mark on the tube, where x=20 mL/(40%/gel percentage %).
CAUTION: Polyacrylamide gels often contain un-polymerized acrylamide which is a toxic material that can produce a hazard when introduced to the environment. Dispose of polyacrylamide gels through the institution’s chemical waste program. - Microwave for 15 s at 30% power. Place on the nutator for 10 min at room temperature or until urea has completely dissolved.
- While the gel mixture is rotating, retrieve a gel cassette (18-well, 1 mm thick; 13.3 x 8.7 cm (W x L)), remove the comb and position the cassette for gel casting.
- Quick spin the 50 mL tube for 5 s to collect gel solution at the bottom of the tube. Add 125 µL of 10% ammonium persulphate solution (APS). Place on the nutator for ~ 1 min. Quick spin again for 5 s.
- Add 25 µL of tetramethylethylenediamine (TEMED) and carefully mix by pipetting up and down 5 times with a 25 mL pipette avoiding bubbles.
- Pipette into the gel cast and carefully insert the gel comb avoiding bubbles.
- Tighten the seal by adding a large binder clip over the top of the cassette.
- Allow the gel to polymerize for 1 h.
- Once the gel has solidified, remove the binder clip. Insert the cassette into the electrophoresis box. Add 1x TBE buffer into the top and bottom reservoirs.
- Retrieve transcription reaction. Add water up to 100 µL, then add 100 µL of 2x Gel Loading Buffer. Prepare the ladder by mixing the recommended amount with water to 10 µL and 10 µL of 2x Gel Loading Buffer to a separate 1.5 mL tube.
- Incubate both the ladder and the in vitro transcription reaction in the thermomixer at 70 °C for 15 min.
- While the samples are denaturing at 70 °C, carefully remove the comb, clean the wells by pipetting up and down each well with a P1000 pipette, and immediately pre-run for 10 min at 100 V.
- Once the samples have completed their 15 min denaturation at 70 °C, remove the tubes from the thermomixer and immediately place on ice.
- Clean each well of the gel again with the P1000 pipette. Load 20 µL of the ladder on the first well to the left, 20 µL of the RNA sample on 10 separate wells, and 20 µL of 1x Gel Loading Buffer on the unutilized wells.
- Run the gel at 100 V for 60 -240 min, depending on the % polyacrylamide of the gel.
NOTE: The dyes in the Gel Loading Buffer will separate into two bands as they traverse the gel, a slow migrating Bromophenol Blue (BB) blue band, and a fast migrating Xylene Cyanol (XC) cyan band. The approximate migration of these bands in different % polyacrylamide gels is well known (see step 1.11) and can be used to estimate the migration of RNA in the gel. - Once the gel has been stopped, carefully remove the gel from the cassette.
- Place the gel in a clean box containing a solution of 50 mL of 1x TBE buffer with 50 µL of nucleic acid gel stain and incubate for 5 min on a rocker to stain the RNA.
- Take a before and after band excision picture of the gel on the gel imaging system, preferably using blue light instead of UV transillumination.
- Excise each band of interest using a nuclease-free disposable gel cutting tip. After each well, transfer the tip containing the gel slice to a 1.5 mL tube and briefly spin to collect the gel slice. Repeat until all the bands have been collected in the same 1.5 mL tube.
- Once all the gel slices have been collected, add 100 µL of nuclease-free water or TE buffer to the 1.5 mL tube. Store at 4 °C for ~48 h. This allows the RNA to exit the gel slices into the solution.
- After 48 h, pipet the water or TE buffer to a fresh 1.5 mL tube. Dispose of the remaining gel slices. Purify the RNA via clean-up kit as follows.
- Equilibrate the spin column at room temperature for at least 30 min.
- To the 100 µL of RNA solution in the new 1.5 mL tube from step 1.32, add 350 µL of RLT buffer and mix well for 2 min on a nutator. Spin for 1 s at 200 x g to collect solution at the bottom of the tube.
- Add 675 µL of 100% EtOH and mix well for 2 min on the nutator. Spin for 1 s at 200 x g and immediately proceed to the next step.
- Transfer 565 µL of the mixture onto the spin column and spin for 1 min at 15,000 x g. Empty the collection tube by aspiration.
- Repeat previous step with the second half of the sample.
- Add 500 µL of RPE buffer to the column and spin for 1 min at 15,000 x g. Empty the collection tube by aspiration.
- Add 750 µL of 80% ethanol to the column and spin for 1 min at 15,000 x g. Empty the collection tube by aspiration.
- Place the column in a new 2 mL collection tube with the lid open and spin at 15,000 x g for 5 min.
- Transfer the column to a new 1.5 mL tube.
- Add 17 µL of water on the center of the column and spin at 15,000 x g for 1 min to elute. Elute again using another 17 µL of water. The total recovered volume should be 32 µL.
- Mix well by vortexing, pipet 1.5 µL and measure the concentration of the RNA using a microvolume spectrophotometer.
- Check the quality of the RNA purification by urea denaturing polyacrylamide gel as in steps 1.11-1.29.
2. In vitro RNA methyltransferase assay
- Set up the 100 µL RNA methyltransferase assay in a 1.5 mL tube on ice as follows: 23 µL of water, 10 µL of 10x TBS (500 mM Tris-HCl, pH 7.5; 1.5 M NaCl), 2 µL of 0.05 M EDTA, 5 µL of 100 mM DTT, 40 µL of 50% glycerol, 4 µL of 58 µM 3H-SAM, 5 µL of 20x Protease Inhibitor Cocktail, 1 µL of RNaseOUT (optional), 5 µL of RNA and 5 µL of Methyltransferase.
CAUTION: Radioactive tritiated material is hazardous and should only be handled while wearing gloves, a lab coat and any other necessary PPE. All pipette tips and tubes in contact with radioactive material are considered solid radioactive waste. Dispose of all solid and liquid radioactive waste according to the lab’s approved radioactive waste protocol.
NOTE: Include control samples without the methyltransferase and without the RNA. The reagent concentrations may require optimization and/or inclusion of divalent cation salts, such as MgCl2, or unlabeled SAM. The optimal range of final RNA concentration is from 50 nM to 1 µM, while the methyltransferase concentration is from 25 nM to 300 nM. - Mix thoroughly by gently vortexing the tube. Spin 5 s at 200 x g to collect the solution at the bottom of the tube. Incubate the tube(s) at 37 °C for 2 h.
- Clean up the reaction using column purification as in step 1.32.
CAUTION: Be very careful to properly dispose of any radioactive materials, particularly during the column washes (pipette out instead of aspirating waste from collection tubes). - Perform liquid scintillation count
- Set up the scintillation count rack with one vial per sample, one vial for background measurement and one vial for the swipe test. Fill the vials with 5 mL of scintillation counting solution.
- Add 10 µL of each eluted radioactive RNA sample into 1 vial, and tighten the lid and mix gently.
- Prepare the swipe test vials. Thoroughly rub cotton swabs on all surfaces and equipment used during the protocol. Add swabs to the vials filled with 5 mL of scintillation solution and tighten the lid.
- Run the samples on the scintillation counter as follows. Open the counter hood, insert the rack into the machine and close the hood. Select Count Single Rack. Select Select User Program. Select or create a program that measures tritium (3H) for 60 s. Hit Count Rack. Repeat the scintillation count three times.
NOTE: The equipment will measure the scintillation count of each sample and output to both the screen and a printout. Protocol can be paused here if desired. Remaining RNA samples from step 2.3 should be frozen at -80 °C for later use.
- Proceed with the autoradiogram
- Prepare and pre-run urea denaturing polyacrylamide gel as in steps 1.11-1.20.
- Pipette 20 µL of radioactive RNA material into a new 1.5 mL tube containing 20 µL of 2x Gel Loading Buffer. Mix well. Prepare the ladder as in step 1.21. Incubate the samples at 70 °C for 15 min.
- Wash the wells of the gel once more immediately before sample loading. Load 20 µL of the prepared ladder, 20 µL of the samples, and 20 µL of 1x Gel Loading Buffer on remaining lanes. Run the gel at 100 V for 60-180 min, depending on polyacrylamide percentage.
- Once the gel finishes running, remove the gel from the cassette and place it in a box containing 50 mL of 1x TBE buffer with 5 µL of ultrasensitive nucleic acid gel stain.
- Incubate for 5 min on the rocker to stain the RNA.
- Carefully take the gel out of the box and place it on the UV transilluminator of the gel imaging system with the wells up and the ladder on the left.
- Focus the camera on the gel, turn on the UV light and then take an image of the gel by exposing from 50 ms to 1 s depending on signal intensity.
- Turn off UV exposure, and save image as Tiff file.
- Place the gel back into the box. Remove TBE. Fix the gel with 50 mL of fixing solution (50% methanol, 10% acetic acid, 40% ultra-pure water) for 30 min at room temperature on a rocker.
- Gently move the gel again to a fresh black box containing 25 mL of the autoradiography enhancing solution. In the absence of a black box, cover the box with aluminum foil to protect the solution from light.
- Incubate for 30 min at room temperature on the rocker.
- Gently lift the gel and place it face-down on a sheet of plastic wrap with the wells up and the ladder on the right-hand side. Place two sheets of chromatography paper on the back of the gel. Gently flip the entire stack.
- Pre-heat the gel dryer to 80 °C. Move back the plastic cover on the gel dryer. Insert wrap, gel and chromatography paper stack under the plastic cover and move the plastic cover back down to create a seal.
- Dry for 1 h at 80 °C in the gel dryer.
- Turn off the gel dryer and gently remove the stack. Remove the wrap and second chromatography paper. Tape remaining chromatography paper with the dried gel in an autoradiogram cassette.
- Add 1 sheet of autoradiography film in the dark room.
- Place the cassette at -80 °C and develop the film after 1 h to 4 weeks depending on signal intensity, which can be judged based on the previously measured scintillation counts: 1-4 weeks for 250-1,000 cpm, 24 h to 1 week for 1,000-10,000 cpm and 1 h to 24 h for >10,000 cpm.
- Once the film is developed, place the film on top of the cassette and carefully mark with a lab marker the 4 edges of the gel, each well, and the position of the XC and BB dyes.
- Scan the film at 300 or 600 pixels per inch resolution and save the image as Tiff file.
Representative Results
In vitro transcription reaction
Figure 2A represents a typical run from an in vitro transcription reaction with the T7 RNA polymerase of the 7SK snRNA, which is a relatively short (331 nt) and highly structured RNA. As shown on that raw image, there are multiple undesired bands, both shorter and longer than 7SK, probably resulting from random transcriptional initiation or termination events. Because of this, gel purification following the in vitro transcription reaction is important to obtain a clean RNA sample as shown in Figure 2B. At this point, the RNA of interest can be identified by its location relative to the ladder and purified by gel purification.
As mentioned previously, it may be necessary to optimize the length of the transcription reaction prior to the gel purification step. Transcription reactions that run for too long can result in extremely high amounts of RNA production, making downstream identification of the correct transcript difficult. It is also important to verify the identity of the purified transcript by Reverse Transcription and quantitative PCR (RTqPCR) using specific primers.
In vitro RNA methyltransferase assay
Figure 3 shows a representative result of an RNA methyltransferase assay described in the protocol using the lower limit of our recommended RNA and protein concentrations. This assay allows for both quantitative results from the scintillation counts, as well as qualitative results from the autoradiogram. Here, MePCE, an RNA methyltransferase known to methylate 7SK10, was shown to be able to also methylate U6. Moreover, as recently shown for 7SK16, binding of histone H4 to MePCE also inhibits U6 methylation. We were able to observe this in both the scintillation count (Figure 3C) and the autoradiogram (Figure 3B), showing the robustness of this protocol.
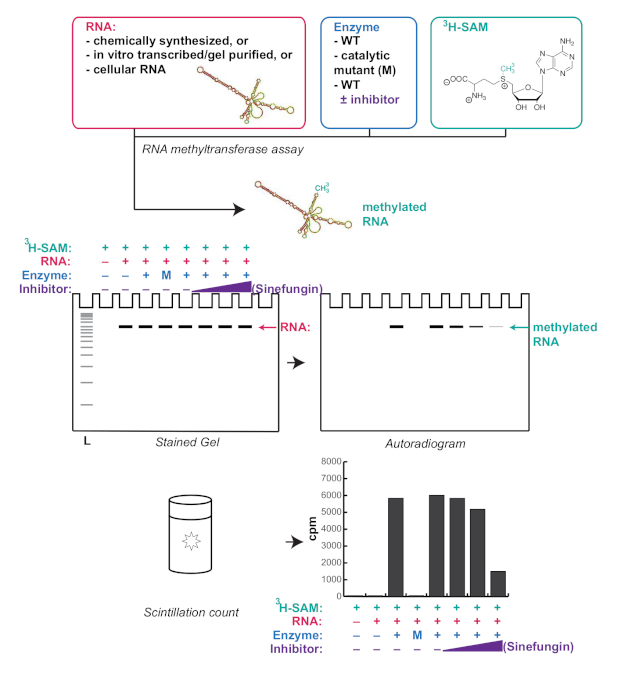
Figure 1. Schematic representation of a typical RNA methyltransferase assay workflow and expected results. Sinefungin is a competitive methyltransferase inhibitor. L: ladder; WT: wild type; M: catalytic mutant; cpm: counts per min. Please click here to view a larger version of this figure.
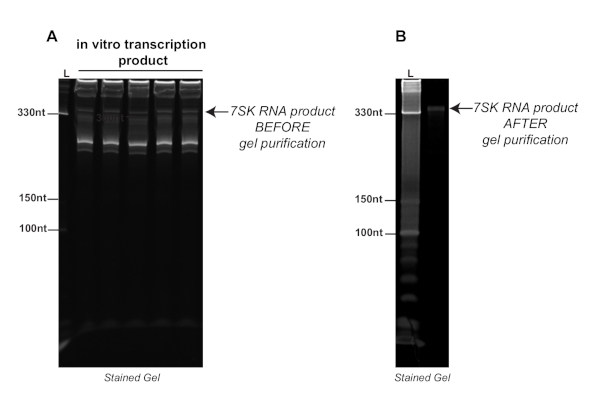
Figure 2. Representative experiment showing the product of in vitro transcription before (A) and after (B) purification on a denaturing urea-polyacrylamide 8% gel stained with nucleic acid stain. The arrows point to the 7SK transcript before and after gel purification. L: ladder. Please click here to view a larger version of this figure.

Figure 3. In vitro RNA methyl-transferase assay performed with MePCE against U6. In vitro methyltransferase assay using recombinant GST-MePCE (25 nM), 3H-radioactive SAM as methyl group donor and in vitro transcribed U6 RNA (50 nM) as substrate. The autoradiogram was exposed for 2 weeks in order to detect the residual activity (250 cpm) in the +Histone H4 sample. Please click here to view a larger version of this figure.
Discussion
Here, a simple and robust method for in vitro verification of RNA methyltransferase activity towards specific transcripts is reported. The assay takes advantage of the fact that S-adenosyl methionine can be tritiated on the methyl group donor (Figure 1), allowing for the methylated RNA to be accurately detected without the use of antibodies. However, it is important to note that this assay cannot indicate which residue or chemical group is methylated by the enzyme. To identify or to verify that a specific residue is methylated, other methods such as mutational analysis, reverse transcription block or mass spectrometry analysis of the RNA substrate can be used in conjunction with the RNA methyltransferase assay.
The choice of RNAs and their mode of synthesis depends on the size of the RNA. Specific RNAs of sizes between 18 and 120 nt can be conveniently custom-synthesized from many reputed nucleic acid providers. However, most commonly, specific RNAs of various sizes are in vitro transcribed. The DNA templates for in vitro transcription can be generated in various ways, and require the sequence of interest to be located downstream of a T7 or SP6 promoter. When precise ends of RNAs are important, the pRZ plasmid is recommended17. The high sensitivity of the assay (Figure 3) also allows to substitute a purified RNA with a mixture of cellular RNA, either total or messenger RNAs for example. Indeed, the gel and autoradiogram provide a method to directly observe the size of the RNA(s) targeted for methylation.
The conditions reported here in step 2.1 of the protocol are optimal for the BIN3 family of methyltransferases, which has two homologs in humans, MePCE10 and BCDIN3D9. It is important to emphasize that the assay conditions need to be adjusted to the specific protein and RNA of interest. For example, it was shown that the presence of MgCl2 decreases BCDIN3D activity18, however, MgCl2 can be an important coordinator of RNA structure, and could thus constitutes an important component of the assay for other RNA methyltransferases.
The advantage of using an autoradiogram, which can be exposed for an extended period of time, is that it can allow to detect very weak activity that is not detected in the scintillation assay. Usually this indicates that the protein is a methyltransferase, but that one or more of the reaction conditions or reagents need to be optimized: enzyme; substrate, cofactor, buffer conditions, etc9. For example, the enzyme purification may need to be improved to remove or change the position of the tag. It may also be that the RNA used as a substrate needs to be folded properly or to interact with another protein or RNA factor to be methylated by the enzyme. Thus, pre-existing literature and/or own results in cells need to be carefully reviewed to set-up the assay conditions for the enzyme and RNA of interest.
The assay is also extremely flexible. For example, it can be a precursor step to another type of assay, such that the radioactively methylated RNA is used in quantitative and qualitative demethylase assays11, or in RNA binding assays allowing to specifically visualize the behavior of the methylated RNA compared to the unmodified one. The assay can also be lightly adjusted to analyze the modification of RNAs that use coenzymes that can be radioactively labeled, such as acetyl coenzyme A for the study of RNA acetylation19,20.
Declarações
The authors have nothing to disclose.
Acknowledgements
The authors would like to thank Dr. Turja Kanti Debnath for his help with ChemDraw. Research in the Xhemalçe lab is supported by the Department Of Defense - Congressionally Directed Medical Research Program - Breast Cancer Breakthrough Award (W81XWH-16-1-0352), NIH Grant R01 GM127802 and start-up funds from the Institute of Cellular and Molecular Biology and the College of Natural Sciences at the University of Texas at Austin, USA.
Materials
| 10 bp DNA Ladder | Invitrogen | 10821-015 | 10 bp DNA Ladder kit. |
| 10% Ammonium Persulfate (APS) | N/A | N/A | For urea denaturing polyacrylamide gel (For 10 mL, dissolve 1g in 8 mL of milliQ water; adjust volume to 10 mL with milliQ water; filter the solution using a 10 mL syringe equiped with a 0.45 µm filter). |
| 10X TBE Buffer | N/A | N/A | For urea denaturing polyacrylamide gel (For 1L, add 108 g of Tris Base, 55 g of Boric Acid to a cylinder with a stir bar; add 800 mL of distilled water and let dissolve; add 40mL of 0.5 M Na2EDTA (pH 8.0); adjust volume to 1L with milliQ water; filter the solution using a 0.22µm filter). |
| 10X TBS | N/A | N/A | For 1L, add 60.5 g of Tris Base, 87.6 g of NaCl to a cylinder with a stir bar; add 800 mL of distilled water and let dissolve; adjust pH to 7.5 with concentrated HCl; adjust volume to 1L with milliQ water; filter the solution using a 0.22 µm filter. |
| Acrylamide: Bis-Acrylamide 29:1 (40% Solution/Electrophoresis), Fisher BioReagents | Fisher | BP1408-1 | For urea denaturing polyacrylamide gel. |
| ADENOSYL-L-METHIONINE, S-[METHYL-3H]; (SAM[3H]) | Perkin Elmer | NET155V250UC | For in vitro methylation of RNA; Concentration = 1.0 mCi/mL; Specific activity = 17.1 Ci/mmol; Molarity= (1.0 Ci/L)/(83.2 Ci/mmol) = 0.0584 mmol/L = 58.4 µM. Upon receipt of the frozen 3H-SAM tube, thaw it at 4°C, make 20 µL aliquots, and freeze them at -30°C. Never refreeze and reuse a partially used aliquot. |
| Amersham Hypercassette Autoradiography Cassettes | GE Healthcare | RPN11649 | For autoradiogram gel exposure. |
| Amersham Hyperfilm MP | GE Healthcare | 28906846 | For autoradiogram gel exposure. |
| Beckman Scintillation Counter | Beckman | LS6500 | For liquid scintillation count. |
| Biorad Mini Horizontal Electrophoresis System | Biorad | 1704466 | Mini Horizontal Electrophoresis System. |
| cOmplete Mini EDTA-free Protease Inhibitor Cocktail Tablets | Roche Applied Science | 4693159001 | For a 20X solution, dissolve 1 tablet in 0.525 mL of nuclease free water. |
| Criterion Cell | Biorad | 345-9902 | RNase free empty cassette for polyacrylamide gel. |
| Criterion empty Cassettes | Biorad | 1656001 | Vertical midi-format electrophoresis cell. |
| DeNovix DS-11 Microvolume Spectrophotometer | DeNovix | DS-11-S | Microvolume Spectrophotometer for measuring DNA and RNA concentration. |
| Ecoscint Original | National Diagnostics | LS-271 | For liquid scintillation count. |
| Fisherbrand 7mL HDPE Scintillation Vials | Fisher | 03-337-1 | For liquid scintillation count. |
| Fluoro-Hance-Quick Acting Autoradiography Enhancer | RPI CORP | 112600 | For autoradiogram gel pretreatment. |
| Gel dryer | Biorad | 1651745 | For drying gel. |
| Gel Loading Buffer II | Ambion | AM8547 | For loading RNA in denaturing polyacrylamide urea gel (composition: 95% Formamide, 18 mM EDTA, and 0.025% SDS, Xylene Cyanol, and Bromophenol Blue). |
| GeneCatcher disposable gel excision tips | Gel Company | NC9431993 | For removing bands from agarose and polyacrylamide gels. |
| Megascript Kit | Ambion | AM1333 | For in vitro transcription with T7 RNA polymerase. |
| Perfectwestern Extralarge Container | Genhunter Corporation | NC9226382 (clear)/ NC9965364 (black) | Gel staining box. |
| pRZ | Addgene | #27663 | Plasmid for producing in vitro transcripts with homogeneous ends |
| Qiagen RNeasy MinElute Cleanup | Qiagen | 74204 | For RNA clean-up, use modified protocol provided in the protocol. |
| QIAquick Gel Extraction Kit (50) | Qiagen | 28704 | Kit for gel extraction and clean up of dsDNA fragment used for in vitro transcription. |
| Saran Premium Plastic Wrap | Saran Wrap | Amazon | For drying gel. |
| SYBR Gold | Invitrogen | S11494 | Ultra sensitive nucleic acid gel stain. |
| SYBR Safe | Invitrogen | S33102 | Nucleic acid gel stain. |
| TE | Sigma | 93283-100ML | 10 mM Tris-HCl, 1 mM disodium EDTA, pH 8.0 |
| TEMED | Fisher | 110-18-9 | For urea denaturing polyacrylamide gel. |
| Thermomixer with SMARTBLOCK 24X 1.5mL TUBES | eppendorf | 5382000023/5361000038 | For temperature controlled incubation of 1.5 mL tubes. |
| TOPO TA Cloning Kit | life technologies | Kits for fast cloning of Taq polymerase–amplified PCR products into vectors containing T7 and/or SP6 promoters for in vitro RNA transcription. | |
| TURBO DNase (2 U⁄µL) | Ambion | AM2238 | For DNA removal from in vitro transcription reactions. |
| Urea | Sigma | 51456-500G | For urea denaturing polyacrylamide gel. |
| Whatman 3MM paper | GE Healthcare | 3030-154 | Chromatography paper for drying gel. |
Referências
- Xhemalce, B. From histones to RNA: role of methylation in cancer. Briefings in Functional Genomics. 12 (3), 244-253 (2013).
- Shelton, S. B., Reinsborough, C., Xhemalce, B. Who Watches the Watchmen: Roles of RNA Modifications in the RNA Interference Pathway. PLoS Genetics. 12 (7), 1006139 (2016).
- Mauer, J., et al. Reversible methylation of m(6)Am in the 5′ cap controls mRNA stability. Nature. 541 (7637), 371-375 (2017).
- Wang, X., et al. N6-methyladenosine-dependent regulation of messenger RNA stability. Nature. 505 (7481), 117-120 (2014).
- Pendleton, K. E., et al. The U6 snRNA m(6)A Methyltransferase METTL16 Regulates SAM Synthetase Intron Retention. Cell. 169 (5), 824-835 (2017).
- Meyer, K. D., et al. 5′ UTR m(6)A Promotes Cap-Independent Translation. Cell. 163 (4), 999-1010 (2015).
- Wang, X., et al. N(6)-methyladenosine Modulates Messenger RNA Translation Efficiency. Cell. 161 (6), 1388-1399 (2015).
- Alarcon, C. R., Lee, H., Goodarzi, H., Halberg, N., Tavazoie, S. F. N6-methyladenosine marks primary microRNAs for processing. Nature. 519 (7544), 482-485 (2015).
- Xhemalce, B., Robson, S. C., Kouzarides, T. Human RNA methyltransferase BCDIN3D regulates microRNA processing. Cell. 151 (2), 278-288 (2012).
- Jeronimo, C., et al. Systematic analysis of the protein interaction network for the human transcription machinery reveals the identity of the 7SK capping enzyme. Molecular Cell. 27 (2), 262-274 (2007).
- Liu, W., et al. Brd4 and JMJD6-associated anti-pause enhancers in regulation of transcriptional pause release. Cell. 155 (7), 1581-1595 (2013).
- JoVE Science Education Database. Basic Methods in Cellular and Molecular Biology. Molecular Cloning. Journal of Visual Experimentation. , (2019).
- Lorenz, T. C. Polymerase chain reaction: basic protocol plus troubleshooting and optimization strategies. Journal of Visual Experimentation. (63), e3998 (2012).
- Rong, M., Durbin, R. K., McAllister, W. T. Template strand switching by T7 RNA polymerase. Journal of Biological Chemistry. 273 (17), 10253-10260 (1998).
- Rio, D. C. Expression and purification of active recombinant T7 RNA polymerase from E. coli. Cold Spring Harb Protocols. 2013 (11), (2013).
- Shelton, S. B., et al. Crosstalk between the RNA Methylation and Histone-Binding Activities of MePCE Regulates P-TEFb Activation on Chromatin. Cell Reports. 22 (6), 1374-1383 (2018).
- Walker, S. C., Avis, J. M., Conn, G. L. General plasmids for producing RNA in vitro transcripts with homogeneous ends. Nucleic Acids Research. 31 (15), 82 (2003).
- Blazer, L. L., et al. A Suite of Biochemical Assays for Screening RNA Methyltransferase BCDIN3D. SLAS Discovery. 22 (1), 32-39 (2017).
- Arango, D., et al. Acetylation of Cytidine in mRNA Promotes Translation Efficiency. Cell. 175 (7), 1872-1886 (2018).
- Ito, S., et al. Human NAT10 is an ATP-dependent RNA acetyltransferase responsible for N4-acetylcytidine formation in 18 S ribosomal RNA (rRNA). Journal of Biological Chemistry. 289 (52), 35724-35730 (2014).
