Neurons in hippocampal CA2 were filled with biocytin following electrophysiological recordings (Figures 1Ac, Ad and 1Bc, Bd). The slices were fixed overnight following the recordings and the neurochemistry and morphological characterization of neurons were revealed following the protocol described here.
The calcium binding protein or protein content of filled interneurons was determined by incubating the slices first with primary antibodies and then with fluorescently labelled secondary antibodies. The firing characteristics of the interneurons during the recordings will dictate the choice of primary antibodies used. Avidin-AMCA was used to visualize the biocytin-filled interneuron and anti-mouse fluorescein isothiocyanate (FITC) and goat anti-rabbit Texas Red (TR) were used to characterize the interneuronal neurochemistry (Figure 1Bb).
Following the fluorescence visualization, an HRP protocol was used to reveal the biocytin (Figure 1Ab). The fine detailed anatomy of CA2 interneurons was then drawn in 3D using a neuron reconstruction software (Figure 2 and Figure 3a). A video of a 3D reconstruction of a basket cell recorded and filled in CA2 is displayed in Video. Neuronal reconstructions were considered as complete if both the dendritic and axonal arbors were confined within the depth of the 450-500 μm slice. Poor axonal arbor staining was assessed by the presence of truncated branches with open endings at the top or the bottom of the slice or by a staining that was limited to the axon initial segment and very proximal branches. Figure 4 represents the examples of a good and a poor HRP staining following biocytin visualization.
Morphometric analysis of 3D reconstructions (Figure 3) can be carried out to demonstrate branch complexity, soma surface area and the surface area and volume of the dendritic and axonal arbors.
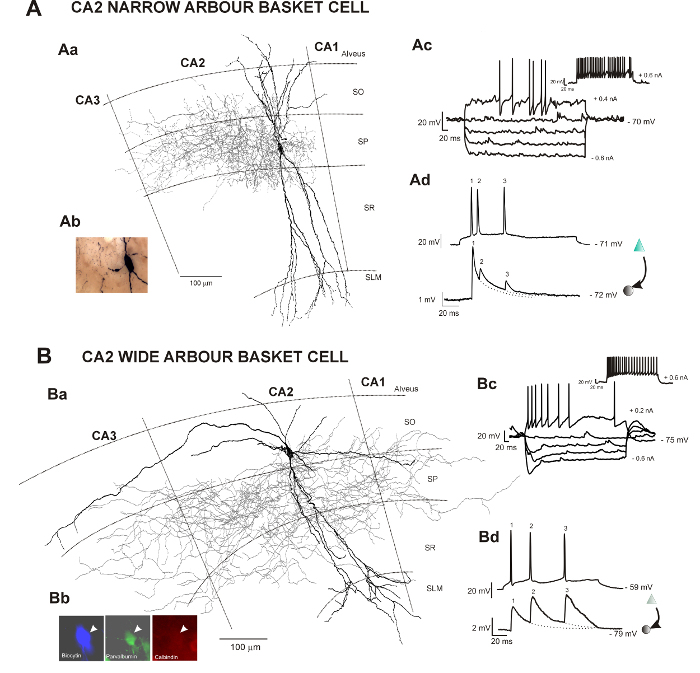
Figure 1: Neuronal reconstructions of two types of basket cells recorded and filled in the hippocampal CA2 region and correlated electrophysiological data obtained following intracellular recordings in vitro. This figure has been modified from previous studies23,24. SO Stratum Oriens, SP Stratum Pyramidale, SR Stratum Radiatum, SLM Stratum Lacunosum Moleculare. (A) Aa: Reconstruction of a CA2 basket cell with restricted dendritic and axonal arbor using a drawing tube (1000X). The dendrites are in black, and the axon is in red. Ab: Image of the biocytin-filled basket cell following the avidin-HRP protocol described here. Ac: Representative trace of voltage responses to hyperpolarizing and depolarizing current injection of a CA2 basket cell with restricted dendritic and axonal arbor. Ad: Example of a CA2 pyramid to narrow arbor basket cell connections recorded using sharp electrodes. Composite excitatory post-synaptic potential (EPSP) averages show brief train depression apparent during responses to trains of three spikes. (B) Ba: 2D reconstruction of a CA2 basket cell with wide dendritic and axonal arbors using a drawing tube (1000X). The dendritic tree of this basket cell (in black) extended radially through all layers of the CA2 region and horizontally in SO and SP of the CA2 and CA3 regions. One horizontal dendrite also reached the CA1 region. The axon (in red) extended to the CA3 and CA1 regions. Bb: The biocytin-filled (AMCA staining) basket cell was PV-immunopositive (FITC staining) and CB-immunonegative (Texas-Red staining). Bc: Representative trace of voltage responses to hyperpolarizing and depolarizing current injection of a CA2 basket cell with wide dendritic and axonal arbor. Bd: Composite EPSP averages show brief train facilitation apparent during responses to trains of three spikes. Examples of other types of interneurons recorded in CA2 can be found in previous studies25,30. Please click here to view a larger version of this figure.

Figure 2: 3D cell body reconstruction. (A) 3D tracing of the cell body. View of the different contours traced at different z positions whilst focusing through the cell body. (B) 3D view of the different contours. (C) 3D view of the cell body at a different angle. Please click here to view a larger version of this figure.
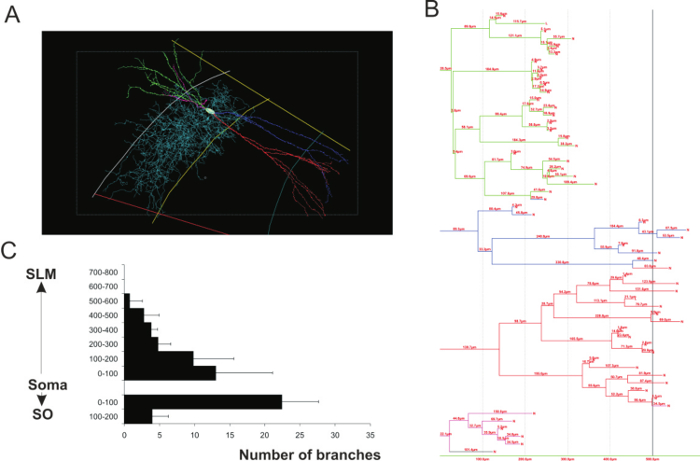
Figure 3: Morphometry analyses of 3D neuronal reconstructions. (A) 3D reconstruction of a CA2 narrow arbor basket cell. Each dendritic branch is represented by a color (green, blue, red and pink) and axon is in light blue. (B) Dendrogram of the basket cell representing the number of dendritic branches and length of each segment contained within spheres concentric with the soma and at 100 μm intervals from the soma. The colors on the dendrogram correspond to those of the dendrites in A. (C) Example of morphometric analysis performed on CA2 pyramidal cells (adapted from a previous study23). The number of dendritic branches was plotted against the distance from the soma of CA2 pyramidal cells. Please click here to view a larger version of this figure.

Figure 4: Examples of good (A) and poor (B) HRP staining. (A) Biocytin recovery revealed a very well filled interneuron in CA2. The cell body (CB) is darkly stained and has clear outlines. The dendrites are beaded and displayed some spines (represented by red stars). The axonal arbor (A) is dense and presents small boutons. (B) Example of a poor dendritic and axonal staining of 2 pyramidal cells in CA2. The staining of the CB is faint with no clear outline. Poor staining is often associated with shorter electrophysiological recordings resulting in the presence of very few biocytin-filled branches. Please click here to view a larger version of this figure.
Video 1: 3D neuronal reconstruction of a CA2 basket cell with restricted dendritic arbor (also referred to as CA2 narrow arbor basket cell) with its soma in stratum pyramidale, dendrites spanning all layers and axon in CA2 stratum pyramidale and adjacent stratum oriens and radiatum. Very few branches reached the proximal CA3 stratum oriens and CA3 stratum pyramidale. This cell was filled with biocytin following electrophysiological recordings and sections were processed with avidin-HRP following the protocol described here. Due to slicing, only the axon within the depth of the slice was recovered, though the dendrites are intact. Dendrites are in dark pink and axon in white. Layer and region boundaries have been added at the beginning of the video. 3D reconstruction by Georgia Economides- 3D video by Svenja Falk. The video was recorded with a neuron reconstruction software as stated in Step 2.17 and edited with a video editing software. Link to the video:30. Please click here to download this video.
| Solutions used | Composition/Instructions |
| Fixation solution | 4% paraformaldehyde, 0.2% saturated picric acid solution, 0.025% glutaraldehyde solution in 0.1 M phosphate buffer (PB) |
| 0.1M Phosphate Buffer pH 7.6 | Add 100 mL of stock 1 M Phosphate Buffer to 900 mL of distilled water |
| Phosphate buffered saline (PBS) pH 7.4/7.5 | Add 10 mL of 0.1M phosphate buffer, 0.2 g of KCl and 8.76 g of NaCl to 990 mL of distilled water |
| TRIS buffer pH 7.5 | Dissolve 5.72 g of Tris Hydrochloride and 1.66 g of Tris Base in 50 mL of distilled water. Then make up to 1 L with distilled water. |
| Buffered glutaraldehyde and paraformaldehyde fixative solution | 4% paraformaldehyde, 0.2% saturated picric acid solution, 0.025% glutaraldehyde solution in 0.1 M Phosphate buffer. |
| ABC solution | Solution to be made at least 30 min before use from the ABC kit. Add 1 drop of solution A and 1 drop of solution B to 2.5 mL of PBS. |
| Durcupan epoxy resin: | To make 20 pots: 20 g of component A, 20 g of component B, 0.6 g of component C and 0.4 g of component D- |
| Protect the balance from spills by covering the plate with a circle of filter paper. Carefully weigh the reagents into a tripour beaker in the proportions stated above. Mix thoroughly by vigorously stirring using two wooden sticks for at least 5 min. The mixture should become a uniform density dark brown colour. Place the beaker into the oven at ~50 °C for a maximum of 10 min to remove as many air bubbles as possible. NOTE: The resin will start to cure if you leave the beaker in the oven longer than 10 min. Decant the resin out into plastic pots or 5 mL syringes, date them and store in the -20 °C freezer ready for use. |
Table 1: Table of solutions.
