Dividing and highly viable cells is an essential starting point for purification of active mitoribosomes. This protocol is applicable to any HEK293 suspension cells. We are using in-house cell line T501, which is stably expressing a transporter under tetracycline-inducible control. The parental cell line is HEK293S-GnTI– cells (Table of Materials)10. During the cell growth and expansion in the FreeStyle 293 Expression Medium the minimal density should be kept at 1.5 x 106 cell/mL, in order to ensure a doubling rate of every two days, whereas the maximal cell density should not exceed ~5 x 106 cell/mL. Upon reaching a cell density above 3 x 106 cell/mL the cells are pelleted and resuspended in a fresh, preheated media in an expanded volume to achieve the minimum cell density of 1.5 x 106 cell/mL. This splitting is performed repeatedly every 2-3 days until a desired cell mass for the procedure is achieved. The final cell density can vary in the range of 3-4.5 x 106 cell/mL, and at least 2 L is required as a starting point for the isolation of mitochondria. The cell viability should be generally maintained >90%, and for the final culture that is harvested >95%. Treatment of the large-scale cell culture with antibiotics is not recommended.
After the series of differential centrifugations, the volume of the mitochondrial suspension after step 2.2.20 is typically in the range of 3-5 mL. The mitochondria are then separated on the sucrose gradient (Figure 1) from other organelles. The stepwise sucrose gradient is prepared such that the volume of the gradient and the volume of the mitochondrial suspension together fill up the centrifugation tube to its maximal volume. Here special care is needed to collect the brown band migrating to the 60% / 32% interface with minimal contamination from the surrounding buffer. This is important in order to keep constant protein:detergent ratio in the following step of mitochondria solubilization. It is advised to assess the mitochondrial protein concentration at this stage, and a typical yield of 15-20 mg total mitochondrial protein is expected from ~1010 cultured HEK cells.
Upon successful mitochondria isolation, they are lysed by the addition of polyethylene glycol octylphenyl ether, and mitoribosomes are separated through a sucrose cushion (Figure 2). To separate mitoribosomes from the hydrophobic membranous substance, pellets are resuspended in the buffer not containing detergent, and hydrophobic complexes are pelleted by centrifugation. This procedure is repeated, and the purification degree of mitoribosomes is quantified by A260/A280 ratio, which is expected to be ~1.3. Typical yield is 7 A260 from a 2 L starting culture. This mitoribosomal fraction also contains additional large soluble mitochondrial complexes, such as pyruvate dehydrogenase and glutamate dehydrogenase. To separate mitoribosomes from the soluble mitochondrial complexes, the supernatant is applied to a sucrose density gradient. Fractions containing the mitoribosome are generally located at the bottom third of the tube. The fractionation of the sucrose gradient can then be done either with an automated piston or manually by carefully taking 50 µL fractions with a pipette or by punching the tube bottom with a 21 G needle and collecting the drops.
Two major mitoribosomal populations are identified in the gradient: monosome 55S and large subunit 39S, as illustrated in Figure 3 and Figure 4. The presence of the large subunit fraction suggests that cells are harvested in a highly active dividing state11. The ratio between the monosome and large subunit peaks may change. An additional peak located closer to the bottom may appear in preparations with contaminating cytoplasmic ribosomes 80S. Please note that the small sucrose gradient using the swinging bucket rotor TLS-55 allows for the rapid purification of ~1 mL of mitoribosomes at an optical density of 0.4-1 A260. The separation of the monosome and large subunit peaks may vary slightly depending on the fractionation, but there is usually some overlap of the two peaks. Hence, which fractions to collect, and pool should be taken into consideration in order to ensure the highest proportion of monosome, or alternatively large subunit, in the sample. For high-resolution cryo-EM studies, the separation between the two mitoribosomal populations is not absolutely required (due to additional in silico operations). However, if better separation is necessary, it is recommended to use larger tubes and corresponding running times.

Figure 1: Purification of mitochondria on a sucrose gradient. Subcellular organelles from a 2 L starting culture were fractionated through a series of differential centrifugations as described in the protocol, and mitochondria were separated on a discontinuous sucrose gradient. The purified mitochondria are found in the lower band at the 32%/60% interface.

Figure 2: Purification of mitoribosomes on a sucrose cushion. Crude mitoribosomes from a 2 L starting culture are sedimented through 1 M sucrose cushion. The pellets are resuspended in a detergent-free buffer, and mitoribosomes are clarified by two centrifugations as described in the protocol. The absorbance is recorded to assess the quality of the preparation and typical A260/A280 ratio of ~1.3 (right panel) certifies a mitoribosome rich fraction. Please click here to view a larger version of this figure.
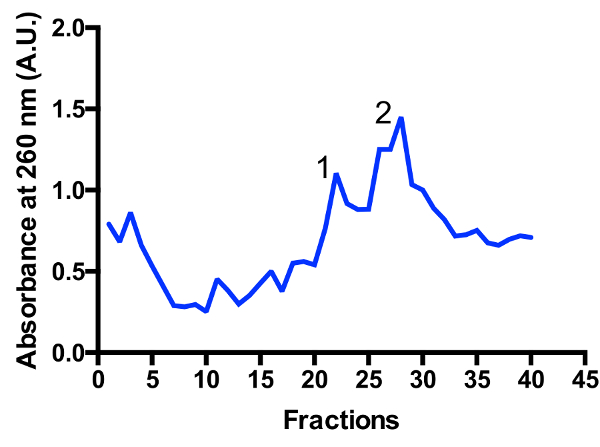
Figure 3: Fine purification of mitoribosomes on a sucrose gradient. Absorbance trace from a 2 L starting culture. Fractions are numbered from the top to the bottom of the gradient. Two major mitoribosomal populations are identified: large subunit (peak 1) and monosome 55S (peak 2). The ratio between the populations may change. Please click here to view a larger version of this figure.

Figure 4: Electron micrograph, 2D classes, and 3D reconstruction. Left panel: a micrograph with the sample from the monosome peak 2 at a calibrated magnification of 1.23 A/pixel. Middle panel: A representative post-processing data (2D classes) revealing intact monosomes. Right panel: 3D reconstruction. Please click here to view a larger version of this figure.
