Figure 1A illustrates first the preparation of DNA-binding beads and quality control using DNA of different sizes (1 kb ladder). 0ne, 2 and 2.5 volumes (1 to 3) of beads was added to one volume of the sample to purify high and low molecular size DNA fragments.
Figures 1 B,C,D are typical examples of DNA gels from whole sonicated DNA extracted from mouse ES cells, embryoid bodies or a cardiac embryonic region (pooled 25 E9.5 ventricles), respectively.
When ES cells differentiate toward a cardiac cell fate, they transit first through a mesendodermal and then a mesodermal state. Such a developmental journey includes a step by which epithelial cells have to undergo an epithelio-mesenchymal transition
Figure 2 shows PCR results following immunoprecipitation of modified histones. The experiment was designed to look at the epigenetic regulation of epithelio-mesenchymal transition genes in wild type or gene-mutated cells when ES cells differentiate toward a mesodermal cell fate. Chromatin from wild type or mutated ES cells treated with a morphogen like BMP2 was immunoprecipitated using anti H3K4me1, H3K36me3 and H3K9me2 antibodies. Binding of modified histones is monitored by PCR of genomic (enhancers, A, B, C, D, E) regions of cadherin and twist genes.
Differentiation genes feature bivalent domains in ES cells12. Their 5' UTR regions close to their promoters are indeed occupied by both H3K4me3 a chromatin activating mark and H3K27me3, a repressed mark. These marks are modified when cells are challenged by morphogens such as BMP2 7. These marks are however variable depending upon the human ES cells lines likely because of the original blastocyst they derive from even when undifferentiated (Figure 3).
The protocols described above are suitable for ChIP sequencing even starting from a low amount of material; ChIP experiments were performed from chromatin extracted from AVC, OFT and ventricles of E9.5 mouse embryos in order to uncover novel genes and enhancers playing a role in defining the identity of these specific cardiac regions. Figure 4 shows 2 genomic regions of myocardial genes in the E9.5 mouse ventricle enriched by an acetylated H3K27 and of one pacemaker-specific (i.e., not ventricular) gene not enriched by acetylated H3K27.
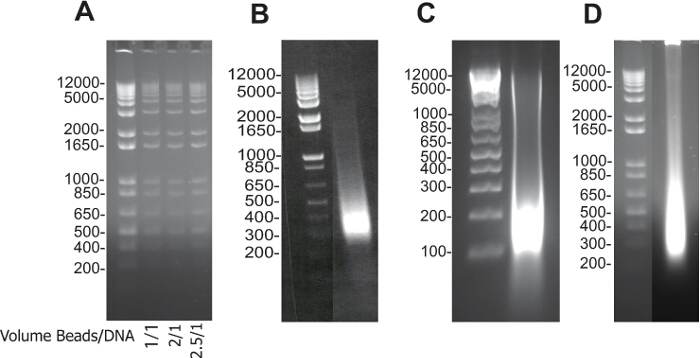
Figure 1. Electrophoresis Data: DNA-binding Beads Checking and Sonication Validation. (A) DNA purified by DNA-binding beads, 1 volume of beads/1 volume of DNA (1) and 2 volumes of beads/1 volume of DNA (2) and 2.5 volume of beads/1 volume of DNA (3). Size of DNA fragments obtained after reverse crosslink of input fractions of mouse ES cells (B), embryoid bodies (C), and embryonic cardiac tissue (D). The samples were run on 2% agarose electrophoresis gels. Please click here to view a larger version of this figure.
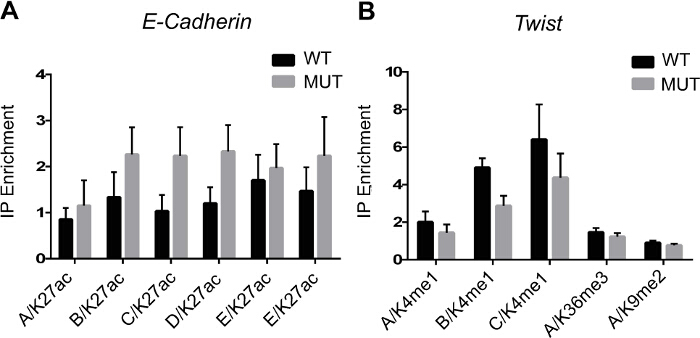
Figure 2. ChIP Data Analysis. (A) Enrichment of H3K27ac epigenetic mark on E-cadherin enhancers/promoter (A - E regions). (B) Enrichment of H3K4me1, H3K36me3 and H3K9me2 epigenetic marks on Twist enhancers/promoter (A-C regions). Genomic regions are amplified by qPCR to measure the histone enrichment vs the input following ChIP. Results are means±SEM from 3 experiments. Please click here to view a larger version of this figure.
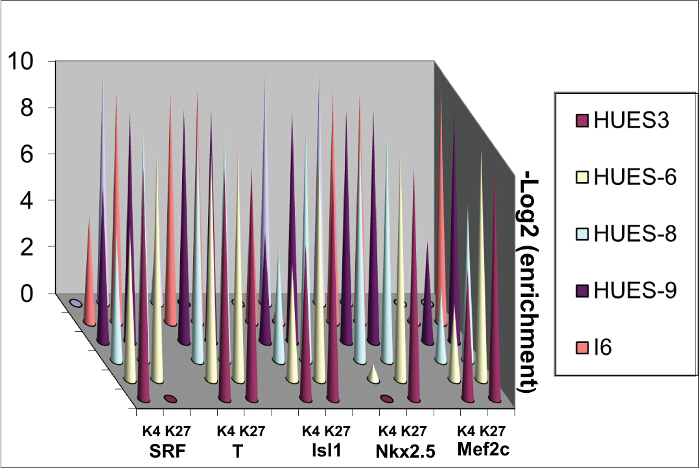
Figure 3. Sequential ChIP. Chromatin was extracted from 4 human ES cell lines and ChIP was performed sequentially using anti H3K4me3 and then anti-H3K27me3 antibodies. Please click here to view a larger version of this figure.
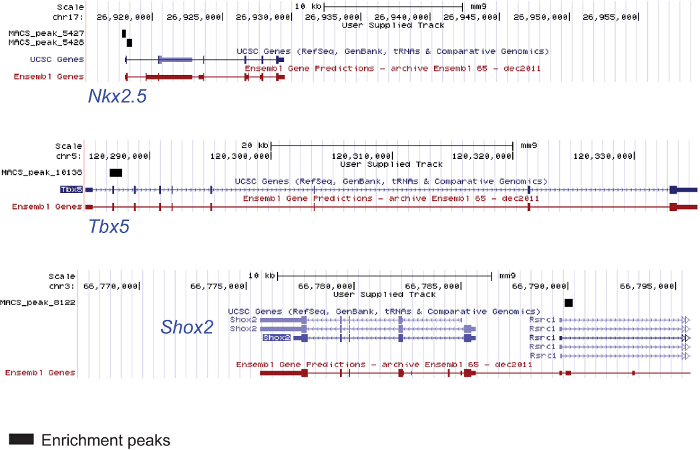
Figure 4. ChIP from Chromatin Extracted from Specific Embryonic Cardiac Regions. Chromatin was extracted from 25 ventricles of E9.5 mouse embryos (2 litters) lysed and sonicated. The anti-H3K27ac antibody was used for immunoprecipitation. The DNA was used for sequencing. The figure shows genomic regions of the cardiac genes Nkx2.5 and Tbx5 enriched in the modified histone and one genomic region of Shox2 pacemaker-specific gene not enriched and not expressed in the ventricle. Please click here to view a larger version of this figure.
