1. Custom ZFN Design Process
- To expedite the ZFN design process, an investigator should provide:
- Information on the target gene such as the gene name, species, and gene annotation/identification number.
- A clearly stated over-all objective of the experiment (e.g., Knockout or Knockin)
- Any specific factors of the gene target which include atypical gene structure, special biology implicated in the genetic locus, or any homologous regions elsewhere in the genome.
- The specific range of DNA sequence for the zinc finger nucleases to target.
- The Sigma bioinformatics team conducts in silico ZFN design to develop initial zinc finger target sites.
- Initial ZFN target sites are provided to a ZFN scientific technical consultant. For more technically complex projects, this Sigma scientist works with the investigators to develop the project strategy.
- The Sigma bioinformatics team conducts an analysis with ZFN algorithms to generate in silico ZFN designs. This includes a whole genome search for off-target sites, repeat masking, and SNP analysis for each potential ZFN target site.
- Bioinformatics provides the top ZFN target sites to the investigator for review. Upon approval of the potential ZFN target sites, this information is provided to the ZFN Production team to begin the ZFN manufacturing process.
2. Custom ZFN Production
- The Sigma archive of zinc finger modules is used to assemble the approved ZFN designs.
- Each ZFN design is assembled and sequence verified on a high throughput cloning platform.
- Once production of all ZFN designs is complete every ZFN is sent to validation.
3. Validation of Custom ZFNs
- ZFN projects in human, mouse, rat or CHO species are validated in well characterized cell lines for each species, a yeast based assay is used for other organisms or cell types.
- ZFN constructs are delivered into the appropriate cell line by nucleofection and the ZFNs are expressed.
- DNA is harvested from the pool of ZFN transfected cells and the region of interest is PCR amplified.
- The Cel-1 assay is then conducted on the PCR product. Gel electrophoresis is run on the Cel-1 assay to validated ZFN activity.
- The highest activity ZFN design as confirmed by the Cel-1 assay is then provided to the customer. ZFNs are delivered in mRNA and plasmid DNA along with PCR primers for the Cel-1 assay and genomic DNA as a positive control.
4. Delivery of Validated ZFNs by Nucleofection
- Seed the cells at a density of 2×105 cells/ml the day before nucleofection.
- On the day of nucleofection, take out Cell Line Nucleofector Kit V and let warm to room temperature.
- Add the supplement to the Nucleofection Solution V according to the manufacturer’s protocol.
- Count the cells. Cell density should be between 2.5-5×105 cells/ml.
- Fill a 6-well plate with 2ml of medium in each well and pre-warm in a CO2 incubator at 37 °C for at least 20 minutes prior to nucleofection.
- Centrifuge 2×106 cells per transfection (8×106 total) at 200xg for 5 minutes.
- Wash cells twice with 20 ml of Hank’s Balanced Salt Solution (HBSS).
- Prepare experimental tubes: (see table in Custom ZFN technical bulletin)
- Remove the 6-well plate containing media from step (6.5) from incubator.
- Resuspend cells in 400 μl (100 μl/reaction) of Nucleofection Solution V.
- One reaction at a time, add 100 μl of cells to each DNA or mRNA-containing tube. Transfer the mixture to a 2 mm electroporation cuvette and nucleofect on a Nucleofector with the appropriate program.
- Immediately after nucleofection of each sample, use a transfer pipette to add ~500 μl of prewarmed medium from the 6-well plate in step (6.9) to the cuvette. Then, carefully transfer cells from the cuvette to the remaining prewarmed medium in the 6-well plate.
- Finish all reactions and return the 6-well plate to the CO2 incubator at 37 °C.
5. Harvesting Genomic DNA after Delivery of ZFNs
- 6-well plate – do not harvest all of the pooled cells. It is important to maintain the culture in order to have cells to single cell dilution clone after you confirm that you have Cel-1 digestion products. One to three days after nucleofection collect the cells to prepare chromosomal DNA using the GenElute Mammalian Genomic DNA Miniprep Kit.
6. Cel-1 Assay
- PCR amplify the genomic DNA from the ZFN transfected samples and the positive control genomic DNA provided in the kit, using the supplied primers. The PCR reaction is set up with the following conditions: (see table in Custom ZFN technical bulletin).
- To generate heteroduplexes from the PCR homoduplexes take 10 μl of PCR reaction from each ZFN treated sample plus the control and use the following program on a thermocycler:
95 °C, 10 minutes
95 °C to 85 °C, -2 °C/second
85 °C to 25 °C, -0.1 °C/second
4 °C, indefinitely - Add 1 μl of enhancer and 1 μl of Nuclease S (from Transgenomic Catalog Number 706025) to each reaction and incubate at 42 °C for 20-40 minutes.
- Run the digestions on a 10% PAGE-TBE gel with proper markers, such as DirectLoad WideRange DNA Ladder (Catalog Number D7058) (see results in Custom ZFN technical bulletin).
- Once positive signal has been detected in ZFN-treated populations by cel-I assay, proceed to clonal isolation and characterization.
7. Clonal Isolation and Characterization
- Single-cell clone the ZFN-treated samples by standard methods including FACS and dilution cloning.
- Allow the clones to expand sufficiently, then split some proportion of cells for genomic DNA harvest, freezing back the remainder as banked material.
- Amplify genomic DNA from the clones using the Cel-I primers and analyze the amplicon by Cel-I assay (with and without WT DNA spiked in), or alternatively use this amplicon to proceed directly to genotyping.
- Genotype candidate clones identified as having edited alleles by the preferred method.
8. Representative Results
An example of the entire CompoZr ZFN workflow is presented in Figure 2. ZFNs are delivered to cells where they bind and cleave the appropriate DNA sequence which creates a double-stranded break (DSB). The natural repair process, non-homologous end joining (NHEJ), repairs the DSB. In some instances aberrant NHEJ results in deletion, insertion or substitution of nucleotides. PCR amplification of harvested genomic DNA results in heteroduplex formation between wild type and modified amplicons after the denaturation/annealing step of the PCR reaction. Addition of the Cel-1 enzyme results in cleavage of any heteroduplex molecules. Cel-1 results are resolved by PAGE analysis to confirm ZFN cleavage. Figure 3 provides a schematic of the Cel-1 assay and the expected results.
Representative Cel-1 results are contained in Figures 4 and 5. Figure 4 contains results from a very active pair of ZFNs (21%) in K562 cells; Figure 5 contains results from a less active pair (2.4%) of ZFNs in K562 cells. ZFN activity is confirmed in both figures by the presence of two PCR fragments below the parental PCR fragment.
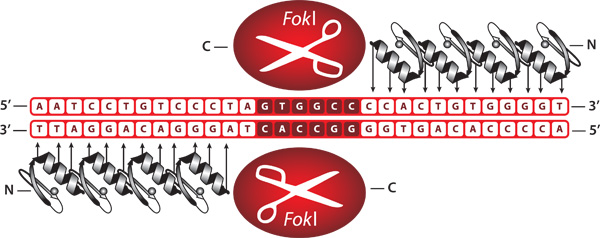
Figure 1. Representation of bound zinc finger nucleases. ZFNs are engineered proteins comprised of a zinc finger DNA-binding domain fused to the cleavage domain of the FokI restriction endonuclease. When bound as a heterodimer, ZFNs create a double-stranded break at a user specified DNA sequence.
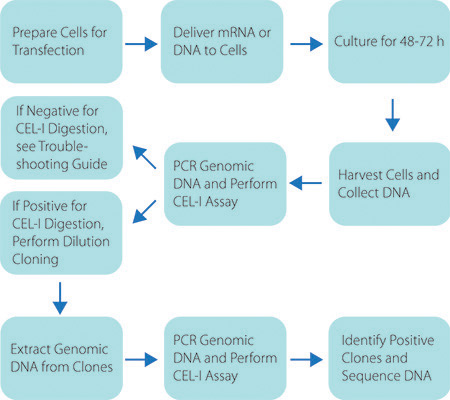
Figure 2. Schematic of the Custom ZFN Service Workflow. Graphic representation of the workflow to generate a genetically modified cell line using CompoZr ZFNs.
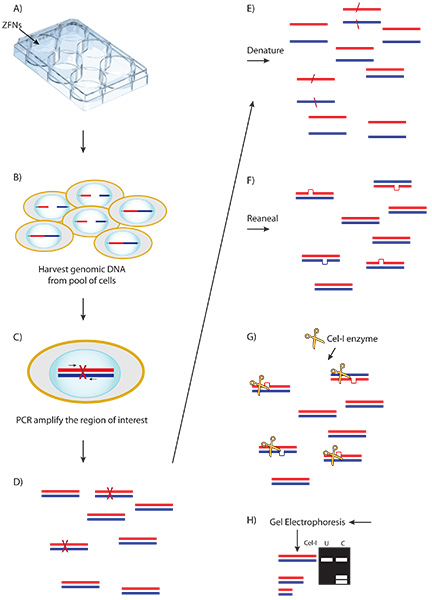
Figure 3. Schematic of the Cel-1 assay and results. A) ZFN plasmid or mRNA is delivered to cells. B) Expressed ZFNs bind and cut their target sequence creating a double stranded break (DSB) in a portion of cells. C) Aberrant repair of some DSBs by non-homologous end joining results in nucleotide insertion or deletion. D) Genomic DNA is harvested from the transfected pool of cells and amplified at the locus of interest. E-F) PCR product is denatured and re-annealed creating heteroduplex formation between wild type and modified amplicons. G) The Cel-1 mismatch endonuclease assay results in cleavage of heteroduplex molecules. H) Cel-1 enzyme digests are resolved by PAGE. The observed ratio of cleavage product to parenteral band, determined by ImageJ software, indicates the fraction cut and efficiency of the ZFNs.
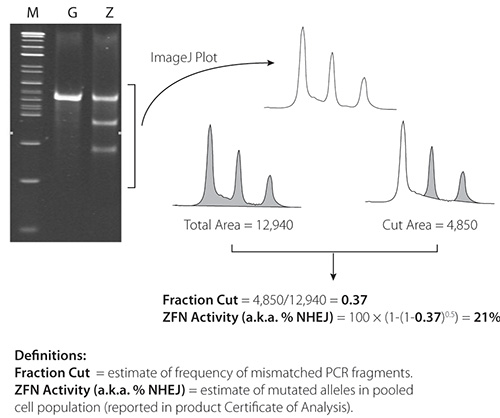
Figure 4. Cel-1 results from 21% active ZFNs. ImageJ software is available at: http://rsbweb.nih.gov/ij/.
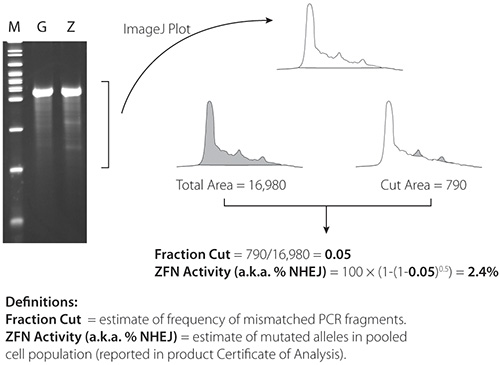
Figure 5. Cel-1 results from 2.4% active ZFNs. ImageJ software is available at: http://rsbweb.nih.gov/ij/.
