Establishing a Whole-Cell Configuration through the Patch Clamp Method
Abstract
Source: Segev, A. et al., Whole-cell Patch-clamp Recordings in Brain Slices. J. Vis. Exp. (2016)
This video demonstrates the whole-cell patch clamp technique in a mouse brain slice. This technique establishes a stable connection between micropipette and neuron membrane, which is further disrupted to establish a whole-cell configuration to study the neuron's health and physiological responses.
Protocol
All procedures involving animal models have been reviewed by the local institutional animal care committee and the JoVE veterinary review board.
1. Slice Preparation
- Construct or obtain a slice recovery chamber.
NOTE: The principle for a recovery chamber is straightforward and can be made in the laboratory (Figure 1). Briefly, the chamber is a receptacle in which a basket is inserted to hold the brain slices at a level that is lower than the surface of artificial cerebrospinal fluid (aCSF). - As an example, obtain four rings (4 – 6 mm high) (Figure 1A, side view; B, top view) by cutting a 30-cc syringe. Then, glue stretched nets (e.g., cut from a nylon hose) to one side of the rings to hold the brain slices (Figure 1B) and glue the rings together.
NOTE: A glue gun can be used.- Once the four rings are glued, glue a curved isosceles trapezoid-shaped plastic wall to two of the rings (Figure 1A and B) to divert oxygen bubbles from the recovering brain slices (Figure 1C and D). As shown in Figure 1D, insert an oxygen diffusing system (here, a gas dispersion tube) on the same side as the plastic walls.
- Prior to slicing, oxygenate (95% O2/5% CO2) and cool down the slicing solution (see step 1.4) to 0 – 2 °C.
- Fill the custom recovery chamber with standard aCSF at room temperature (RT). Ensure that the aCSF is well oxygenated (20 – 30 min, time may vary according to the chamber volume) before placing slices in the recovery chamber. Ensure that gas bubbles do not come in direct contact with the slices or disrupt them.
- Line the vibratome ice tray with ice and fill it with cold water so that one-third to one-half of the slicing chamber is submerged. Carefully place an oxygen delivery system (e.g., gas diffusing stone) and a temperature probe in the slicing chamber so neither item interferes with the blade movement or slice manipulation.
- Prepare the dissection area and tools necessary for extracting the brain and dissecting the desired brain region.
NOTE: The exact dissection performed will depend on the specific brain region studied as different brain structures will require slicing at different planes (e.g., coronal, sagittal, or horizontal slices).- Place the following tools on an underpad: decapitation scissors, scalpel, small straight sharp tip scissors, vessel cannulation forceps (or any surgical tool with a wide tip, such as rongeurs, which is more suitable for rat skulls), curved hemostatic forceps, tweezers, spatula, scooping spatula, filter paper, Petri dish, single edge razor blade, and cyanoacrylate glue.
- When temperature reaches 0-2°C, transfer the slicing solution to the slicing chamber (buffer tray).
- Anesthetize the mouse in a desiccation chamber using isoflurane. The exact amount may vary according to the size of the chamber used, but for a small shoebox cage, use a few drops (~3-4). Leave the mouse in the cage until rendered immobile (not responding to tactile stimuli; around 15 sec for the conditions described here). Perform tail and foot pinch tests to ensure the animal is deeply anesthetized, then decapitate before the heart stops beating (enhances cell viability).
NOTE: With appropriate justification, some laboratories obtain the authorization to perform live decapitation in order to minimize as much as possible excitotoxic processes and enhance cell viability. - Perform the dissection.
NOTE: The brain must be extracted rapidly (<45 sec).- Using the scalpel cut the superficial skin on top of the skull from rostral to caudal.
- Peel the scalp on each side of the head.
- Using small straight sharp tip scissors, cut the interparietal plate along the lambdoid suture to remove the cerebellum. Remove the occipital bone.
- Using the same scissors, cut the sagittal suture.
- Slide the vessel cannulation forceps (or rongeurs if breaking a rat skull) below each parietal bone and pull to expose the brain.
- Using the curved hemostatic forceps, pinch the frontal bones to break them, then use tweezers or vessel cannulation forceps to remove the broken bones. Cut and remove the dura mater as gently as possible, as it can interfere with the dissection.
- Slide the spatula below the brain and gently pull it out of the skull. Place it in the slicing chamber (buffer tray) previously filled with ice-cold aCSF. Let the brain cool down for 1-2 min.
- Prepare the dissection platform by filling a Petri dish with ice and some ice water to allow greater surface contact. Cover the dish with its lid and place filter paper on top. Wet the filter paper with cold aCSF.
- Once the brain is cooled down, place the brain on the ice-filled petri dish and quickly perform the appropriate dissection to obtain the desired plane of slicing.
- To obtain sagittal slices containing the nucleus accumbens (NAc), use a single-edge razor blade to cut and remove the olfactory tubercles and the cerebellum if they are still present. Then, perform a sagittal cut of 2-3 mm from the lateral border of the right hemisphere to obtain the flat surface that will be glued on the specimen holding plate (see step 1.8.11).
NOTE: Cutting only 2 – 3 mm from the lateral border of the hemisphere will allow the collection of slices containing the NAc from both hemispheres. The appropriate dissection will depend on the brain region that is investigated. Here, the dissection is performed so NAc neurons can be recorded in sagittal brain slices. - Rapidly glue (using cyanoacrylate glue applied to the specimen holding plate) the flat cut surface of the brain onto the plate according to the desired plane of slicing. To obtain sagittal brain slices see step 1.8.10.
- Immediately place and secure the specimen holding plate in the slicing chamber so the brain is sliced rostro-caudally (for safety, set up the blade holder only when the specimen plate is secured).
- Set the vibratome with appropriate slicing parameters (parameters used in the lab for the vibratome mentioned in Materials: speed 3 – 4, vibration 9-10, and slice thickness 250 µm).
- Upon slicing, use a plastic-trimmed transfer pipette to transfer the brain slices to the recovery chamber (at RT) (see step 1.3). Recovery time may vary depending on the neuronal type that is under study (typically 30 – 90 min).
2. Recording Micropipettes and Rig Preparation
- Refer to the specific guidelines of the puller user's manual to obtain the desired micropipette properties.
NOTE: For MSNs, we use a pipette resistance range of 3.2-4.0 MΩ. - Oxygenate the aCSF and adjust the flow to 2 ml/min. Vacuum aCSF using a peristaltic pump or vacuum lines installed in the facility.
- Turn on the perfusion heater controller and adjust the temperature settings in order to obtain the desired temperature (e.g., 31.8-32.2 °C).
NOTE: Temperature stability depends upon having both a constant aCSF level and constant flow velocity in the chamber. Since several biophysical properties of neurons (e.g., input resistance, Ri, also called membrane resistance, Rm) are temperature-sensitive, maintaining a stable temperature is important. - Turn on computer-controlled amplifier, camera, micromanipulator, and microscope background light. If performing an experiment that requires electrical stimulation of the tissue, turn on stimulus controller and the isolation unit.
NOTE: Some amplifiers from other manufactures recommend a "warm-up" before use, so it is recommended to consult the manual for the exact operating procedure. - Start camera capture, signal acquisition and amplifier software.
- Slice Placement and Visualization:
- Using a plastic trimmed-tip transfer pipette, gently draw in one brain slice from the recovery chamber.
- Place the transfer pipette in the recording chamber and gently squeeze the slice out of the pipette onto the coverslip lining the bottom of the chamber.
NOTE: As long as no overflow is occurring, it is harmless to have some aCSF from the recovery chamber spilling into the bath. - Use forceps to alter the position of the slice so the desired area will be placed exactly in the center of the recording chamber. Use the microscope low power (4X) objective lens and the eyepiece for assistance in positioning.
- After the desired position has been achieved, secure the brain slice position with a slice hold-down (also known as a "harp") in the chamber.
- Switch to high power (40X) objective lens and lower it gently until contact is formed with the aCSF in the chamber.
- Use the fine adjustment wheel to bring the tissue into focus. While in contact with the aCSF, do not use the coarse adjustment wheel on the microscope as lowering the objective lens excessively can crush the slice or even break the cover slip lining the bottom of the chamber, which can cause aCSF to spill onto the condenser and damage it.
- When the focus is at tissue level, observe cells in the targeted region for shape. Dead cells are easily identifiable by their swelled plasma membrane and nucleus (Figure 1E). Healthy cells should appear as round, ovoid, or elliptical homogenous structures (Figure 1E).
- Look for a target cell. Mark it on the computer screen in order to help guide the recording micropipette. If using software such as QCapture, draw a square around the target cell by holding the left mouse click.
- Raise the objective lens so there will be sufficient space in the cone formed by the objective lens being in contact with the aCSF to place and move the recording micropipette.
- Micropipette Placement and Positioning
- Using a 1 ml syringe, a nonmetallic microsyringe needle, and a dedicated filter, fill a micropipette with the internal solution prepared in advance according to the planned experiment (K+-based or Cs+-based internal solution, see Materials for composition). Use enough solution so the internal solution comes into contact with the chloride-coated silver wire electrode within the micropipette holder.
NOTE: The silver wire electrode can be chlorinated by soaking it in household bleach. Nucleoside triphosphates (adenosine triphosphate, ATP & guanosine triphosphate, GTP) can be added to the internal solution prior to use. Keep the syringe containing the solution on ice to prevent ATP/GTP degradation. - Make sure there are no air bubbles in the micropipette as they can come out while the micropipette is in the tissue and obscure the slice.
- Place the micropipette in the electrode holder so the solution comes in contact with the silver chloride coated wire electrode.
- Tighten the pipette cap so that the cone washer will form a seal around the micropipette.
- Apply positive pressure before immersing the micropipette in the aCSF to prevent debris from entering the pipette.
- Place the headstage in the locked position (facing the chamber), and using the micromanipulator, guide it down towards the chamber so it is roughly under the center of the immersed objective.
- While moving the micropipette with the micromanipulator (set at medium to high speed), use the computer screen to locate the micropipette and guide it toward the location of the cell on the X-Y axis.
- Measure the micropipette resistance by applying a voltage step (e.g., 4 mV for 100 msec), which can be accomplished manually or automatically via specific software such as 'bath' mode if using "Membrane Test" in Clampex software (see also step 4). In order to make sure no air bubbles or any other foreign objects block the micropipette, apply positive pressure using the air-filled syringe (e.g., 30 cc syringe) connected to the micropipette holder with polyethylene tubing.
- After clearing the micropipette, perform a voltage offset to reduce pipette current to zero, which can be accomplished manually or via specific software such as 'pipette offset' on the computer-controlled amplifier commander.
NOTE: This function will compensate for any voltage caused by concentration differences between the bath and the micropipette solutions (i.e., liquid junction potential).
- Using a 1 ml syringe, a nonmetallic microsyringe needle, and a dedicated filter, fill a micropipette with the internal solution prepared in advance according to the planned experiment (K+-based or Cs+-based internal solution, see Materials for composition). Use enough solution so the internal solution comes into contact with the chloride-coated silver wire electrode within the micropipette holder.
3. Membrane Test
NOTE: This step applies to the amplifier mentioned in the Materials.
- When using a computer-controlled amplifier commander, always set it on voltage-clamp mode to perform the membrane test.
NOTE: When membrane test is set in "Bath" mode, the membrane test allows the measurement of the micropipette resistance and the seal resistance when the seal is formed. - Once the membrane is ruptured (see step 4.8), switch the membrane test to "Cell" mode so that series resistance (Rs) (also called access resistance, Ra), Ri and membrane capacitance (Cp) can be obtained.
4. Final Approach, Seal Formation, and Obtaining the Whole-cell Configuration
- Using the fine focus wheel, start focusing down while lowering the micropipette gradually. Always focus down first and then lower the micropipette down to the plane of focus. This will ensure that the micropipette tip will not abruptly penetrate into the slice.
- When the micropipette comes in touch with the surface of the slice, slow down the micromanipulator speed to medium-low mode.
- Gently apply light positive pressure with the air-filled syringe connected to the pipette holder to clear any debris from the approach path.
- Approach the cell either by alternating with the X-Y-Z control knobs, or by approaching diagonally (if the micromanipulator model allows it) where both X-Z axes are changed with the rotation of the Z axis knob. The latter method will prevent vertical compression of tissue.
NOTE: Here, the goal is to approach the cell by inflicting minimal damage to the slice. When the micropipette is close enough to the cell a dimple appears (a round discoloring of the cell surface caused by the positive pressure applied through the tip of the micropipette) (Figure 2). - When the dimple appears (Figure 2-1), apply a weak and brief suction through the tube that is connected to the pipette holder suction tube in order to create the seal (Figure 2-2). Keep monitoring the membrane test.
NOTE: If a partial seal is formed (<1 GΩ), injecting negative currents by lowering the holding potential (on the computer-controlled amplifier commander) can facilitate seal formation and reach gigaohms resistance ("gigaohm seal" or "gigaseal" >1 – 5 GΩ). The high resistance of the seal (>1 GΩ) will both limit noise contamination to the recorded signal and contribute to the mechanical stability of the patch. - While a gigaseal is forming, use the computer-controlled amplifier commander to bring the cell's holding potential as close as possible to physiological resting potential (Vrest) in order to prevent sudden changes once the membrane is ruptured. For example, MSNs are usually voltage-clamped at -70 or -80 mV (physiological Vrest: -70 to -90 mV).
- After the gigaseal has formed, compensate for fast and slow capacitance manually or automatically. If using a computer-controlled amplifier commander such as Multiclamp commander, press 'Auto' for 'Cp Fast' and 'Cp Slow".
- If the seal remains stable and above 1 GΩ (or injecting less than 10 – 20 pA to hold the cell at the desired membrane potential), apply a brief and strong suction through the same tube as in 4.5 to rupture the plasma membrane (Figure 2-3).
NOTE: This might take several trials. A good membrane rupture is achieved when suction is performed strongly enough so that ruptured membrane does not clog the micropipette (which may lead to an increase in Rs during recording), but weakly enough in order to not draw in a large portion of the membrane or the cell. - After achieving a successful whole-cell configuration, regularly monitor the micropipette location to assess and correct for significant drift as it may lead to loss of the patch. Drift amplitude may vary according to several factors, e.g., the quality of the rig installation and pulling forces on the headstage. Ideally, drift should be almost non-existent.
- By switching to "Cell" mode in the membrane test, view different parameters of the cell such as Ri, Rs and Cp. Monitor these parameters during recording.
NOTE: All these parameters can help assess initial health status of the cells and cell types (see "Membrane test" section, step 4).
Representative Results
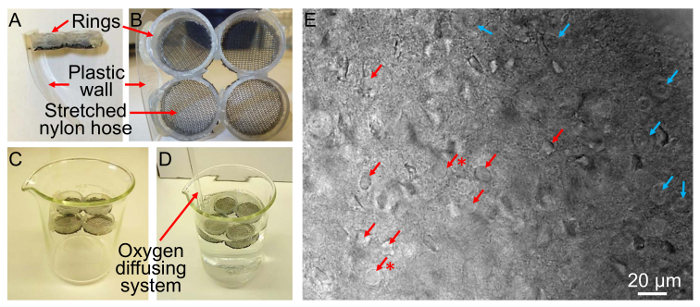
Figure 1. Custom-made Recovery Chamber (A-D) and a Picture of a Brain Slice at 400X Showing Healthy and Dead Neurons (E). A-D) The procedure to make a custom recovery chamber is described in step 2.1. E) Picture of NAc medial shell MSNs in a brain slice at 400X showing examples of healthy (red arrows) vs. dead neurons (blue arrows). Note that although some cells are indicated as healthy, their spherical aspect indicates that they may not be as healthy as desired (red arrows with asterisks). Final health status is assessed based on Vrest and Ri after achieving whole-cell configuration.
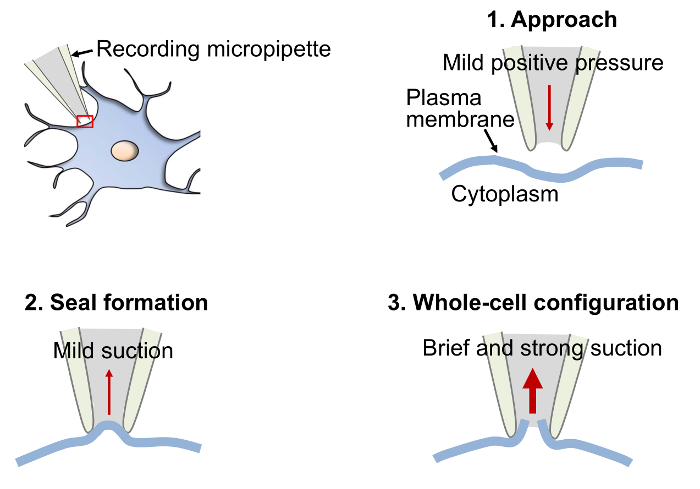
Figure 2. Diagram Depicting the Basic Procedural Steps to Obtain a Gigaseal and Establish the Whole-cell Configuration. When the micropipette is close enough to the cell to create a dimple in the plasma membrane (step 1, Approach), apply a brief and gentle suction to create a tight contact between the micropipette and the plasma membrane. If performed properly, the contact will strengthen and the resistance will increase and reach 1 GΩ (gigaseal) or more (step 2, Seal formation). Once the seal is stable and above 1 GΩ, apply a brief and strong suction to rupture the plasma membrane (step 3, Whole-cell configuration). Achieving the whole cell configuration will allow continuity between the cytoplasm and the micropipette interior. For details, see protocol step 4.1-4.8.
Declarações
The authors have nothing to disclose.
Materials
| Isolated pulse stimulus generator | A.M.P.I | Master-8 | |
| Isolation unit (ISO-Flex) | A.M.P.I | ISO-Flex | |
| Computer controlled Amplifier | Molecular Devices | Multiclamp 700B | |
| Digital Acquisition system | Molecular Devices | Digidata 1500 | |
| Microscope | Olympus | BX-51 | |
| Micromanipulator | Sutter Instruments | MPC-200 | |
| Chamber and in-line Heater | Warner Instruments | TC-344B | |
| Vibratome Slicer | Leica | VT1000 S | |
| Micropipette Puller | Narishige | PC-10 | |
| Imaging Camera | Q Imaging | QIClick-F-M-12 | |
| Narishige pipette puller PC-10 | Narishige | PC-10 | |
| Glass capillaries | WPI | TW150F-3 | |
| Slice hold-down (harp) | Warner Instruments | 64-0255 | |
| Slice Chamber | Warner Instruments | RC-26 | |
| Nonmetallic syringe needle | World Precision Instruments | MF28G67-5 | |
| Syringe filters | Nalgene | 176-0045 | |
| Glue Gun | Home Depot | various | |
| Gas dispersion tube | Ace Glass Inc. | various | |
| Decapitation scissors | Home Depot | 100649198 | |
| Scalpel Handle #3 | World Precision Instruments | 500236 | |
| Small straight sharp tips scissors | World Precision Instruments | 14218 | |
| Vessel canulation forceps | World Precision Instruments | 500453 | |
| Curved hemostatic forceps | World Precision Instruments | 501288 | |
| Economy Tweezers #3 | World Precision Instruments | 501976-6 | |
| Spatula | Fisher Scientific | 14357Q | |
| Scooping spatula | Fisher Scientific | 14-357Q | |
| Petri dish | Fisher Scientific | 08-747B | |
| Filter paper | Lab Depot | CFP1-110 | |
| Solutions | |||
| K-Gluconate internal solution (pH 7.2–7.3, 280–290 mOsm) | |||
| K D-gluconate, 120 mM | Sigma Aldrich/various | G4500 | |
| KCl, 20 mM | Sigma Aldrich/various | P3911 | |
| HEPES, 10 mM | Sigma Aldrich/various | H3375 | |
| EGTA, 0.2 mM | Sigma Aldrich/various | E4378 | |
| MgCl2 | Sigma Aldrich/various | M8266 | |
| Na2GTP, 0.3 mM | Sigma Aldrich/various | G8877 | |
| MgATP, 2 mM | Sigma Aldrich/various | A9187 | |
| Standard artificial cerebrospinal fluid (ACSF, osmolarity ≈ 300-310 mOsm) | |||
| KCl, 2.5 mM | Sigma Aldrich/various | P3911 | |
| NaCl, 119 mM | Sigma Aldrich/various | S7653 | |
| NaH2PO4-H2O, 1 mM | Sigma Aldrich/various | S9638 | |
| NaHCO3, 26.2 mM | Sigma Aldrich/various | S8875 | |
| Glucose, 11 mM | Sigma Aldrich/various | G8270 | |
| MgSO4-7H2O, 1.3 mM | Sigma Aldrich/various | 230391 | |
| CaCl2-2H2O, 2.5 mM | Sigma Aldrich/various | C3881 | |
| Additional compounds used for solutions preparation | |||
| KOH | various | ||
| Kynurenic acid | Sigma Aldrich/various | K3375 |
