Handling Neuronal Stem Cell Cultures in a Cell Production Facility
Abstract
Source: Stover, A. E., et al,. Culturing human pluripotent and neural stem cells in an enclosed cell culture system for basic and preclinical research. J. Vis. Exp. (2016).
The video outlines the procedure for maintaining a controlled environment in a cell culture workstation for the growth and maintenance of neuronal stem cells. These procedures involve setting chamber conditions, cleaning protocols, and media preparation, Thus, maintaining maximum sterility and reproducibility and replacing the traditional methods.
Protocol
1. Initial Setup
- Setting Gas and Temperature Concentrations
- Using the software, click the "Guest" tab on the upper left corner of the graphical interface. Log in to the system using a designated username and password. Make sure each user has their username and password.
- Click on a module to adjust (Figure 1). Within the new window displaying the current settings, click on the existing O2 set point value below "Set point" and enter this module's required O2 concentration level. Enter 5% O2 if pluripotent stem cells will be grown or manipulated in this module and 9% O2 if neural stem cells will be grown or manipulated. Click on the green check mark to confirm the set point.
Note: Two modules are not gas-adjustable: the laminar hood and the microscope chamber. The gas concentrations in the laminar flow hood are atmospheric, while those in the process chamber passively maintain those in the microscope chamber. - Repeat this step for CO2. Enter a set point of 5% CO2 for all modules except for the buffer chambers, which only adjust for O2.
- Monitor the current gas concentrations labeled "process values" to ensure they reach the new set points.
- Adjust the gas set points of all modules to the appropriate values. Match the CO2 and O2 levels of chambers that will be exposed to one another. For example, adjust the process chamber to 5% O2 before opening an incubator that grows cells at 5% O2. Also, change to 5% O2 in any buffer chamber used to add items to the process chamber during this time.
- Set the temperature of the process chamber to 37 °C using the same screen for gas adjustments. Also, the process chamber floor temperature should be set to 37 °C.
- Click on the incubation module banks underneath each incubator. Adjust the temperature of the banks to 37 °C.
- Operation of Buffer Chambers
- Gather all the supplies required for the given task (feeding, splitting, staining, etc.) in the beginning to prevent a delay in the workflow. Liberally spray all materials with 70% ethanol and allow them to dry in the laminar hood. Do not spray flasks or plates of cells—wipe them gently with a sterile gauze sponge saturated with 70% ethanol.
- Ensure the outer and inner doors of the buffer chamber are securely closed. Then, open the outside door and place the items inside.
- Close the outer door. Select the buffer module within the software and click on the dilution factor tab. Enter one into the log factor box. Click start.
Note: A "dilution factor" means that the buffer module's atmosphere will be evacuated and replaced once with clean, HEPA-filtered gas. - Once the graphical interface stops flashing "dilution factor," open the buffer chamber's inner door and bring the materials inside the process chamber.
Caution: Do not allow the inner and outer doors ever to be open simultaneously, and do not open the inner door if the buffer chamber has not undergone a dilution factor. - First, remove items from the process chamber and ensure that the buffer chamber has undergone a dilution factor. Then, open the inner door, place the items to be removed inside, and close the inner door. Then, open the outer door and remove the items.
Note: A dilution factor must not be run before opening the outer door.
2. Introducing and Feeding Cells
- Incubator Setup
- Place 3 Petri dishes in the water pan at the base of each incubator. Fill these dishes with sterile water, not directly filling the pan. Maintain the water level in these dishes, as they are critical for maintaining the incubators' relative humidity (RH) set point.
- Within the graphical interface, click on the incubator. Under the set point, click on the existing relative humidity (RH) value and enter 85%. Click on the green check mark to accept this value.
- Preparation of the Cell Production Facility for Cells
- If not already done so, adjust the buffer chambers, the process chamber, and an incubator to the gas set points required for the type of cell being introduced.
- Clean the surface of the process chamber with sterile gauze and a non-flammable disinfectant. Do not use any peracetic acid-based disinfectant; in a closed system, the strong, lingering vapors are toxic to mammalian cells. Instead, use benzalkonium chloride-based products.
- Continue disinfecting. Clean the gloves and surfaces commonly touched, such as door handles. Use the disinfectant thoroughly but sparingly, as excess liquid in the process chamber will contribute to humidity, condensation, and possible microbial growth.
- Clean the surface of the laminar flow hood and buffer chamber with 70% ethanol and allow it to dry. Place the flask or plate of cells (in our case, PSCs(pluripotent stem cells) or NSCs(neural stem cells)) in the hood and briefly wipe its exterior surface with a sterile gauze sponge saturated with ethanol.
- Put the flask or plate in the buffer chamber and run a dilution factor (step 1.2.3).
- Upon completion of the dilution factor, immediately move the flask or plate to the appropriate incubator. As the incubators are at the back of the process chamber, pull a shelf out into the process chamber for cell placement. Avoid opening the incubator doors unnecessarily or for extended periods, as the process chamber's air is very dry compared to that of the incubators and will result in a significant loss of humidity in the incubator.
- Feeding Cell Cultures
- Gather medium components, such as serological pipettes, an electronic pipettor, gauze, and disinfectant. Also, gather a waste container for the spent cell culture medium, but do not fill it with bleach. Bring the materials into the process chamber through a buffer chamber as described in Section 1.2. Arrange materials to enable an optimal working space. Avoid placing items not being used in the center of the process chamber.
- Leave the media container slightly uncapped to allow the medium to equilibrate with the CO2 and O2 levels present within the system. Some PSC medium manufacturers indicate not to warm media at 37 ºC; if so, use a buffer chamber to equilibrate the medium. Allow the medium to equilibrate for 20 min.
- Remove the old medium from wells using an appropriate stereological pipette and an electronic pipettor. For PSCs, remove all but a small remaining amount of the old medium to prevent desiccation during the feeding process. For NSCs, remove half of the old medium. Pipet the waste into the waste container.
- Place the used pipette back into its original wrapper and move it to the side of the process chamber or into a buffer chamber designated for waste removal. With a fresh sterile serological pipette, add fresh medium to cell culture plates and place the plates back into their designated incubator.
- At the end of feeding, spray gauze with disinfectant and thoroughly clean the floor and door handles of the process chamber. If multiple cell lines are being grown in the system, sterilize between feeding each cell line. This helps minimize the risk of cross-contamination.
- Remove waste or other unneeded items through one of the buffer chambers upon completion.
3. Splitting Cell Cultures
- Enzymatic Passaging of PSCs or NSCs
- Gather all items needed for passaging. This includes media, enzyme or non-enzymatic cell dissociation buffer (the latter is for NSCs only), DPBS (Dulbecco's Phosphate Buffered Saline), plates or flasks, extracellular matrix (ECM), conical tubes, and serological pipettes. Bring them into the system using a buffer chamber.
- Coat fresh plates or flasks with ECM, as previously described 9,10. Some ECMs -such as those derived from mouse sarcoma cell lines – are only liquid between 4 and 15 °C, so use a sterilized cold block or gel ice pack to keep the ECM cold while working in the heated process chamber.
- Pipet off the spent medium from the culture and dispose of it in the waste container.
- Rinse the well or flask surface using 1 ml of DPBS/ well and dispose of the DPBS.
- Add 1 – 2 ml of warm enzyme to each well. Only very dense cultures require 2 ml.
- Immediately move the culture dish or flask to the microscope chamber and observe the culture. Watch for signs of individual cells beginning to loosen off the dish. Do not wait until the cells float into suspension before proceeding to the next step.
- Return the cells to the main process chamber. Using a 5 ml serological pipette, add 4 ml of DPBS for each 1 ml of enzyme, and then forcefully pipet up and down to dislodge the cells from the well surface. If passaging multiple wells within a multiwall plate, add the DPBS to each well before dislodging the cells from the individual wells.
- Transfer the enzyme and PBS cell suspension to an appropriately sized conical tube.
- If a centrifuge is unavailable in the cell production facility, tightly seal the conical tube, place it in a buffer chamber, and seal the door closed.
- Remove the conical tube from the buffer chamber from the laminar flow side and spin the cells at 100 x g for 5 min at RT.
- Spray the conical tube with 70% ethanol and bring it into the process chamber using a buffer chamber and dilution factor.
- Pipet off the supernatant and resuspend the cells in 2 ml of PSC or NSC medium. If passaging PSCs, include ROCK(Rho-associated protein kinase) inhibitor in the medium at a concentration of 10 µM.
- Count the cells using a hemocytometer and determine the wells or plates required. Plate PSCs at 5 x 104 – 1 x 105 cells/cm2. Split the NSCs at a 1:2 ratio.
Note: NSCs often clump and resist accurate counting in a hemocytometer. - If cryopreservation of the cells is required, ensure that all supplies and freezing media are prepared and placed inside the process chamber. DMSO(Dimethylsulfoxide) is very toxic to cells at 37 ºC, so it works quickly. Keep the isopropanol-jacketed freezing container at RT in the laminar flow hood. Once the cryopreservation vials are filled and sealed, immediately remove them from the process chamber by passing them through a buffer chamber.
- Manual Passaging of PSCs
- Confirm that the culture needs to be passed, as previously described. If so, prepare fresh plates or flasks coated with extracellular matrix. Then, change the medium entirely.
- Clean the microscope chamber with sterile gauze moistened with a sparing amount of non-flammable disinfectant—too much will result in excess fogging. Only clean the gloves, floor area, and stage. Bring several 200 µl or 1,000 µl individually wrapped pipet tips into the chamber.
- Bring the plate of PSCs into the microscope chamber, remove the lid, and place it face down on the freshly cleaned floor. Leaving the lid up exposes the underside directly to air currents and potential contamination.
- Unwrap a pipet tip, and using the microscope to visualize the culture, pick apart colonies with good morphology using the tip of the pipet.
Note: The pipet tip may be attached to a pipet if the individual user finds this more comfortable. - Replace the lid on the plate and move the plate back to the process chamber.
- Pipet off excess ECM from the new plate and transfer the media from the old plate (containing the colony pieces in suspension) to the new plate. If the old plate still contains colonies that are not ready to be picked, feed them as well.
Representative Results
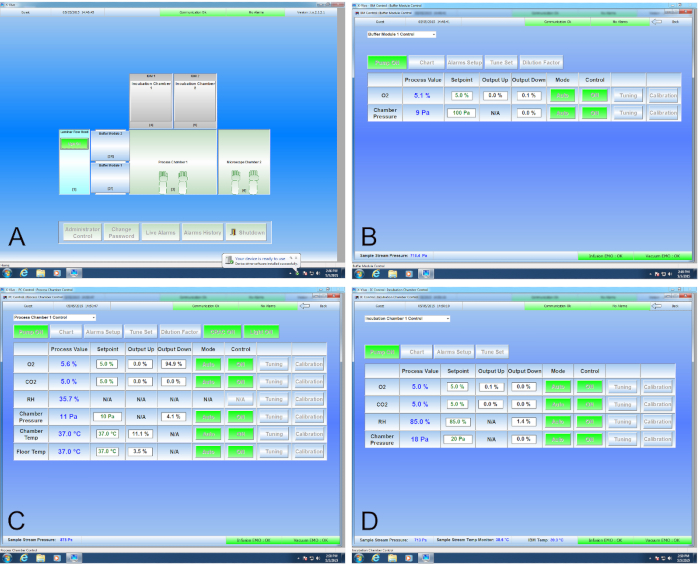
Figure 1. Graphical Interface for the Cell Production Facility. (A) The graphical representation of the CPF is the default screen and matches the CAD drawing shown in Figure 3. A mouse click over Buffer Module 1 opens its control screen (B), where O2 values are set to match the processing chamber. Similarly, clicking the Process Chamber or Incubator 1 brings up their respective control screens (C and D, respectively), where values may be changed or monitored as appropriate.
Declarações
The authors have nothing to disclose.
Materials
| Xvivo System | Biospherix | custom made | |
| Xvivo Software | Biospherix | version i.o.2.1.2.1 | |
| O2 Manifold | Amico | P-M2H-C3-S-U-OXY | |
| CO2 Manifold | Amico | M2H-C3-D-U-CO2 | |
| N2 Manifold | Western Innovator | CTM75-7-2-2-BM | |
| O2 Gas | |||
| CO2 Gas | |||
| N2 Gas | |||
| Petri dish | |||
| Water | |||
| Sterile gauze | |||
| 70% ethanol | BDH | BDH1164-4LP | |
| benzalkonium chloride-based disinfectant | |||
| Dulbecco's Phosphate Buffered Saline | |||
| Hank's-based Cell dissociation Buffer | Life Technologies | 13150-016 | |
| 2mL Serological pipet | VWR | 89130-894 | |
| 5mL Serological pipet | Olympus Plastics | 12-102 | |
| 10mL Serological pipet | Olympus Plastics | 12-104 | |
| 25mL Serological pipet | Olympus Plastics | 12-106 | |
| 50mL Serological pipet | Olympus Plastics | 12-107 | |
| 6-well plate | Corning | 353046 | |
| 20uL pipet tips | Eppendorf | 22491130 | |
| 200uL pipet tips | Eppendorf | 22491148 | |
| 1000 pipet tips | Eppendorf | 22491156 | |
| Microscope with DP21 camera and fluorescence | Olympus Corporation | CKX41 | |
| Phosphate-Buffered Sodium | Hyclone | 9236 | |
| Dimethyl sulfoxide | Protide | PP1130 | |
| Gloves |
