This article provides a method for culturing and exposing human bronchial epithelial cells at the ALI that mimics realistic, repeated inhalation exposure conditions that can be used for toxicity testing. Characteristics of both the cell model and of the exposure system are essential for achieving a realistic inhalation exposure model that can be used for repeated exposures. Results on these characteristics are shown below.
Cell model requirements and selection
When selecting a suitable cell model, the following characteristics must be taken into account:
- The cell model should be able to form a confluent monolayer with functioning tight junctions to mimic the lung barrier.
- The cell model should show optimal performance when exposed repeatedly to conditioned (temperature and humidity) air.
- The cell model should respond to an exposure.
This study started with four different human bronchial epithelial cell lines: 16HBE, Calu-3, H292, and BEAS-2B. These are all widely used for toxicity testing of nanomaterials and chemicals. Of the four cell lines, only the Calu-3 cells fulfilled all the above requirements. The cells formed a monolayer with tight junctions (Figure 3) that remained a stable barrier over time as measured by TEER, whereas the other cell lines either did not form a barrier or showed a drop in barrier function when cultured at the ALI (Figure 4). In addition, H292 and BEAS-2B tended to overgrow into multiple cell layers when cultured for a longer time period. Traditional submerged cell culturing and ALI culturing differed greatly, because at the ALI nutrients were only available from the basolateral side and the cells were exposed to dry conditions at the apical side. These conditions can cause stress to the cell models, which could be observed by measuring the cell viability over time. Cell lines 16HBE, H292, and BEAS-2B all showed an increased LDH release when cultured at the ALI, while Calu-3 cells showed only a slight LDH release (Figure 5).
Next, the response of the Calu-3 model to substances was tested. As a positive control substance, LPS was administered via nebulization to the apical side of the model. The deposited dose was 0.25 µg/cm2. The Calu-3 cells showed a reaction to lipopolysaccharide (LPS) by an increase in LDH release and in tumor necrosis factor alpha (TNF-α) release (Figure 6).
Finally, the Calu-3 monolayer was exposed to quartz silica (DQ12) nanomaterials (IOM, Edinburgh). Crystalline silica can induce silicosis and may also cause lung tumors. Therefore, the International Agency for Research on Cancer (IARC) has classified crystalline silica as a Group I human carcinogen20. The mechanism of action of crystalline silica is thought to be via the induction of persistent inflammation caused by its reactive surface21,22,23. Several in vivo studies in both rats and mice report the induction of inflammation and histopathology changes, including tumors and fibrosis, after inhalation exposure to crystalline silica24,25,26,27,28,29. These effects are all observed after repeated exposure and/or long-term follow-up. The Calu-3 model was used to investigate whether the observations from the in vivo studies could be mimicked using an in vitro model that could be exposed repeatedly at the ALI.
Calu-3 cells were exposed for 3 consecutive weeks, 5 days per week, 4 h per day to DQ12. The deposited dose was measured using a QCM. The average deposited dose was 120 ng/cm2 per day, with a cumulative dose of 1.6 µg/cm2, similar to the doses inducing an effect in vivo. Other particle characteristics are shown in Table 1. After 3 weeks of exposure, DQ12 induced no significant effects in TEER, cell viability, and monocyte chemoattractant protein 1 (MCP-1) release, compared to the clean air controls (Figure 7). As more toxicity of DQ12 was expected based on the in vivo data, the reactivity of the particles was checked using an acellular assay according to the protocol optimized within the EU-project GRACIOUS (deliverable 5.3). The reactivity of the DQ12 batch was lower than expected (Figure 8), orders of magnitudes lower compared to the positive control particles carbon black (CB). This lack of reactivity might explain the absence of a toxicity response in the Calu-3 model.

Figure 1: The automated exposure station (AES).
The left figure shows the outside of the cabinet with the touch panel. The AES has three levels with exposure modules: the top level for clean air exposures, and the middle and bottom level for aerosol exposures. The right figure shows the exposure module in which inserts with cells are placed. Please click here to view a larger version of this figure.
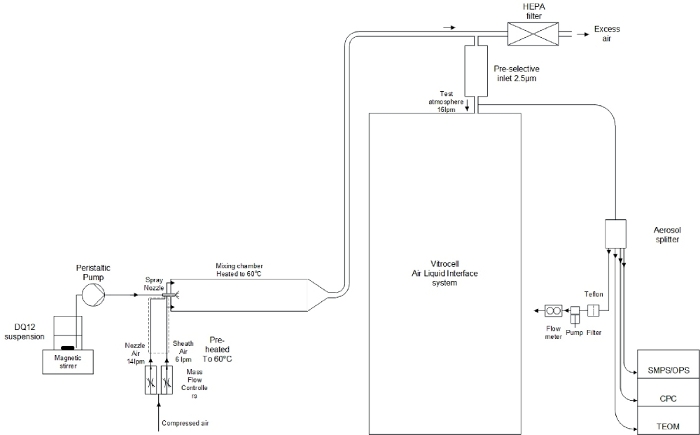
Figure 2: Schematic representation of the exposure setup.
From left to right: 1) the ENM suspension connected to the spray nozzle via a peristaltic pump; 2) Using compressed air the spray nozzle nebulizes the ENM suspension and via a mixing chamber the aerosols are led to the AES; 3) Just before entering the AES, aerosol characterization instruments are connected: SMPS, OPS, CPC, and TEOM. Please click here to view a larger version of this figure.
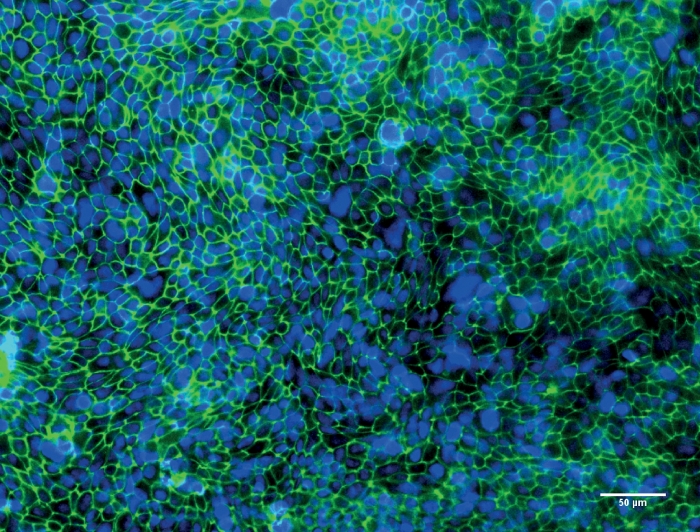
Figure 3: Representative image of Calu-3 cells after culturing at the air-liquid interface (ALI) for 10 days.
Fluorescence microscopy image of Calu-3 cells after culturing at the ALI for 10 days. Tight junction protein ZO-1 is stained in green, nuclei of the cells are stained in blue. Please click here to view a larger version of this figure.
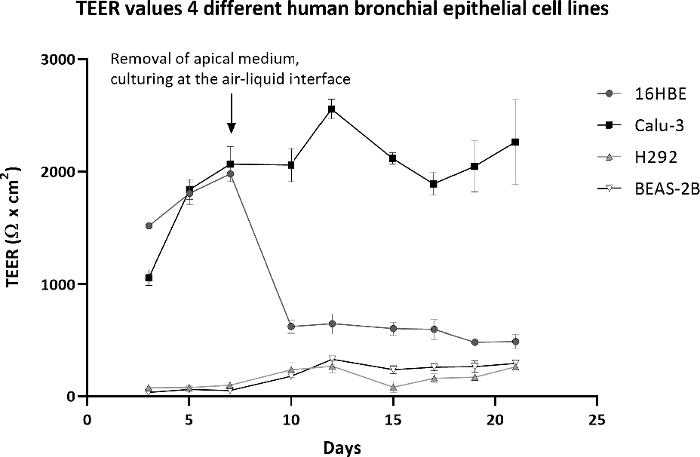
Figure 4: Transepithelial electrical resistance (TEER) of four different cell lines during a culture period of 21 days.
TEER values of 16HBE, Calu-3, H292, and BEAS-2B when cultured for 21 days: first 7 days submerged, followed by 14 days at the ALI. TEER values were corrected for the background resistance of the insert and multiplied by the surface area of the insert. The symbols and error bars represent the average value and standard deviation of six inserts. Please click here to view a larger version of this figure.
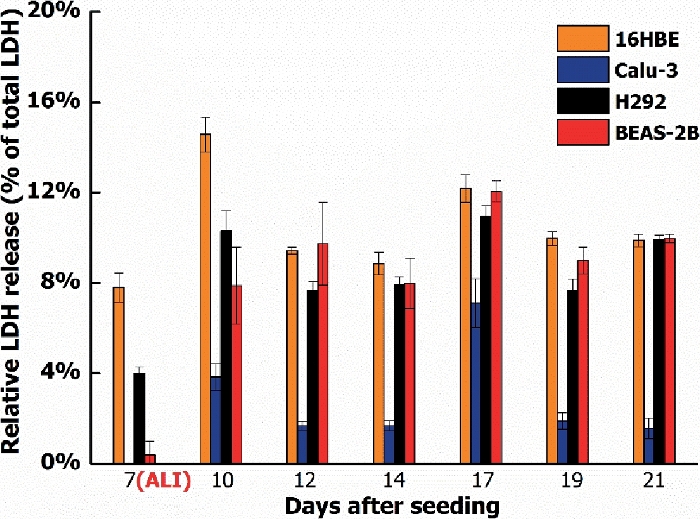
Figure 5: LDH release of four different cell lines during a culture period of 21 days.
LDH release of 16HBE, Calu-3, H292, and BEAS-2B when cultured for 21 days: 7 days submerged, followed by 14 days at the ALI. LDH values shown are relative to the maximum LDH release per cell type. The symbols and error bars represent the average value and standard deviation of five inserts. Please click here to view a larger version of this figure.
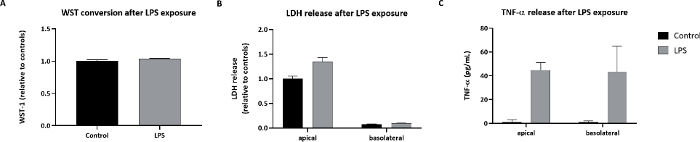
Figure 6: Cellular effects in Calu-3 cells exposed to LPS.
Calu-3 cells were exposed via cloud nebulization to 0.25 µg/cm2 LPS. (A) The WST-1 conversion. (B) LDH release. (C) TNF-α release after LPS exposure. The symbols and error bars represent average values and standard deviations of three inserts. Please click here to view a larger version of this figure.
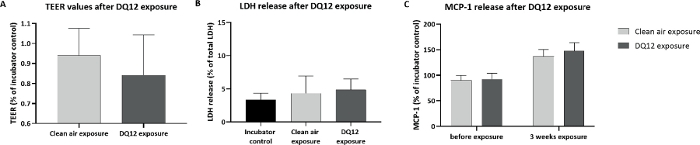
Figure 7: Cellular effects in Calu-3 cells exposed to DQ12 nanomaterials.
Calu-3 cells were exposed for 3 weeks (4 h per day, 5 days per week) to DQ12 nanomaterials, about 120 ng/cm2 per day, cumulative dose of 1.6 µg/cm2. (A) TEER values. (B) LDH release. (C) MCP-1 release after DQ12 exposure. All symbols and error bars represent average values and standard deviations of three inserts for the controls and six inserts for the DQ12 exposure. Please click here to view a larger version of this figure.
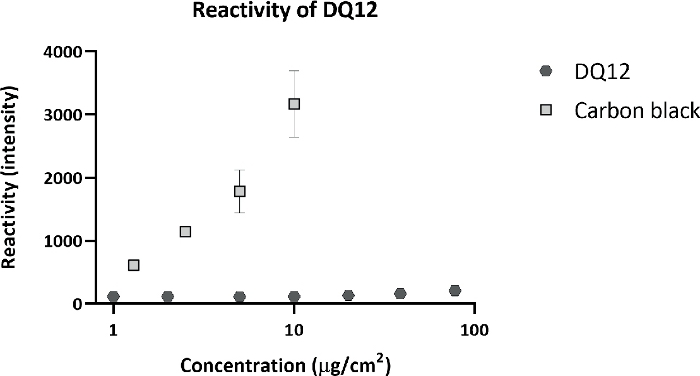
Figure 8: Acellular reactivity of DQ12.
DQ12 was incubated with a 2ʹ,7ʹ-Dichlorofluorescein Diacetate (DCFH-DA) probe to detects its surface reactivity. As a positive control, carbon black (CB) particles were included. Compared to CB, DQ12 has very low surface reactivity. Please click here to view a larger version of this figure.
| Particle mass (µg/m3) | Particle number (#/cm3) | Mobility particle size (nm) | Geometric standard deviation | Optical particle size (µm) |
| 2969 (418) | 83983 (10215) | 66.6 | 2.5 | 1.1 (1.3) |
Table 1: DQ12 exposure characteristics. Values are shown as average with standard deviation in brackets.
