Using phage display technology, the scFv gene anti-EGFR was created from the mouse B-cell hybridoma line C3A820,21. The single chain fragment variable (scFv) structure models of the VH and VL structures were built separately, according to Chua et al.22. After that, the models were visible as ribbons produced using RasMol. Then, using molecular modeling software, a synthetic peptide [Gly4Ser)3 was used to join the separately modeled VH and VL structures. The ligand interaction with the receptor is triggered as a local structure that changes the binding sites called variable ends. The complementarity-determining region (CDR) found in antibody protein structures) creates a natural grooving binding for epitope binding3,23. Antigenic binding causes a CDR conformational change to fit residue 183 with the ideal shape and facilitate the proper physicochemical properties of the residues. Instead of comparing the crystal structures of the antibody-bound and antibody-unbound molecules, a dynamic examination of the binding process is thought to be the best way to understand the underlying allosteric 3D mechanism23.
As shown in Figure 1A-D, the plot is used to determine whether the conformation of the backbone is correctly predicted. As shown in Figure 1A,C, most of the peptides are in the correct center of the plot, demonstrating the accuracy and correctness of the structures in Figures 1B,D. The models are subsequently evaluated using Ramachandran plots to determine the accuracy of these models24. The full scFv model-building and evaluation process were prepared so that the average energy profile was below zero. The two Ramachandran plots showed the correct backbone confirmation for the receptor template of 3njp and scFv antibody template of 1osq. The majority of peptides are centralized β sheets24, while the rest are in the α helix of the plot25. The Ramachandran plot is the fastest and easiest method to detect structural peptide correction and accuracy24,26.
Figure 2A shows the crystal structure of the binding interaction between the receptor and ligand in the surface structure. Figure 2B shows the interaction profile of confirmation and lowest binding. Protein-protein docking was performed with PyRx software, and the ligand was kept flexible while the protein structure was kept rigid. The macromolecule and the ligand were prepared with the aid of the MGL 1.5.7 tools program. The ligand was synthesized by rotation to make it flexible, and the macromolecule was created by removing water molecules and adding hydrogen atoms. Antibody-bound and antibody-unbound 186 crystal structures were used to store the potential energy arising from the interaction between each atom in the flexible ligand13. Within the functional domain of the chosen protein molecule, the ligand was docked around the discovered conserved region16. The preferred interaction was selected based on the favorable binding conformation between the macromolecule and ligand that had the lowest binding energy (B.E.), as determined by the docking program25,26. The grid box size was 40 Å x 40 Å x 40 Å along the X, Y, and Z axes and centered at 90.653, 56.2181, and 50.1986, whereas the targeted docking grid box size was set to 40 Å x 40 Å x 40 Å and centered at the X, Y, and Z coordinates of 49.086, 25.0, and 25.0, respectively. The size of the grid box was generated manually.
Figure 3A shows the ligand-receptor residue interaction (receptor interaction). As shown, the dark green residues show the residues that had stronger binding between the scFv antibody-EGFR (1ivo) with H-bonds. In contrast with the red residues with different types of bonding, as shown in Figure3A, the total number of residues is 1117, defined as A=513, B=510, C=7, and D =447. A is the receptor, there are 8685 heavy atoms, and the interaction charges are +55. Furthermore, eight hydrogen bond interactions are shown in Figure 3B: 1H-bond interaction of DNA. Protein with asn:32 of protein receptor, 1 H-bond interaction of DNA. protein with asn:33 of protein receptor, 4 H-bonds with 3tpo, 1 H-bond with tp 3p45, and 1H-bond with t3p5627. Therefore, these interactions exhibit very good docked complex stability and strength.
As shown in Figure 3B, the hydrogen bonds for docking stability (ligand-receptor interaction) and the number of detected hydrogen bonds always determine the strength of docking stability. Protein folding is made more likely by hydrogen bonds, and the chain has access to all degrees of freedom to achieve its ideal structure. However, for stiff binding, the protein molecules are already folded27,28,29 , leaving the chains with just six degrees of freedom in translation and rotation to arrange themselves into the most advantageous bound configuration.
Molecular dynamics simulations (MDS) of EGF receptor binding with scFv protein were observed. In Table 1, counter ion/salt information, applying the NaCl method to EGF receptor binding with the scFv protein binding interaction, and root-mean-square deviation (RMSD) can be used to detect regions of metastable states in addition to the stable state. The authors observed that the predicted structures are not stable, and there is a possibility that the trajectory obtained by a simulation is not reliable. Therefore, the MD simulation can show the consistent movement pattern of the trajectory obtained from the docking of the receptor-antibody interaction, resulting in a steering force that produces stable values in the predicted structures.
As shown in Table 1, the computations are carried out with NaCl concentrations ranging from C = 0.5 m to C = 0.75 m. Analyses are performed on how ions affect the hydrogen258-linked water network and the tetrahedral structure30. Because of a non-tetra-dihedral configuration, the analysis demonstrates that increasing salt content lowers the tetrahedral arrangement of water31. We identify the origin and angular properties of this nontetrahedral structure using the nearest neighbor method. Furthermore, the tetrahedral structure of water is more significantly impacted by the comparison ions Na+ and Cl– than by other ions30,31. The RMSD approach is the standard method. In the RMSD, the protein is located on the Y-axis. The protein frames and the structural backbone's reference frame frequently line up. When hydrogen bonds are estimated in close proximity to the most stable structure of the Trp-cage, the RMSD is calculated32,33.
As shown in Figure 4, the root mean square deviation (RMSD) was calculated; the protein RMSD increased gradually from 3.2 Å to 6 Å for the first 0 to 20 ns time step. Then, from 20 to 75 ns, the protein RMSD was maintained in the range 4 Å to 5.6 Å and then changed from 75 ns to 90 ns. Finally, the protein RMSD movements increased sharply until 7.2 Å and decreased until 6.4 Å up to the last timestep. This indicates the free movement of the proteins. This shows the instability of the docked structure when the time step increases, and the stability of the receptor-ligand docked structure is provided when the time step of RMSD is in the range of 20 to 75 ns. The above plot shows that the RMSD evaluates the structural conformation throughout the simulation. RMSD analysis indicated that the simulation was balanced and stable.
Root mean square fluctuation (RMSF), is shown in Figure 5. In the RMSF, a particular atom's displacement was compared to a reference structure, and the average number of atoms was measured. It is frequently used to discern whether a structure is stable in the time scale of the simulations or not. Furthermore, in Figure 5, the protein shows an RMSF in the range of 2.4 Å for residues between 1-100. Then, it decreases to 2a for 200-300 residues; furthermore, it fluctuates to 5.6 Å or 500 residues and then sharply declines to 2.4 Å for residues until 1000 ns. Finally, there is one sharp peak at the 1000 position with a decrease until 2.4 Å at the terminal. The RMSF results help characterize changes in the ligand atom positions during docking25. These results represent the binding process, and usually, the final model, the closest model, represents the crystal structure in terms of RMF, and binding interactions can be shown clearly in final images, as demonstrated in the previous study by Huang et al.2.
As observed in34, the docked structure of the EGFR-scFv antibody was stable over 1000 ns of MDsimulation. Therefore, protein secondary structure elements (SSEs), such as alpha helices and beta-strands, were monitored throughout the simulation to observe the changes in the residue index in the formation of protein SSEs35,36. Assignment over time is shown in Figure 6. Protein secondary structure elements (SSEs), such as α-helices (Figure 6 in red) and β-strands (Figure 6 in blue), are monitored throughout the simulation. The plot above reports the SSE distribution by residue index throughout the protein structure. As shown in Figure 7, the cartoon model shows protein segments, lipids, and water molecules arranged in straight lines and the lipids' phosphorus atoms in beads.
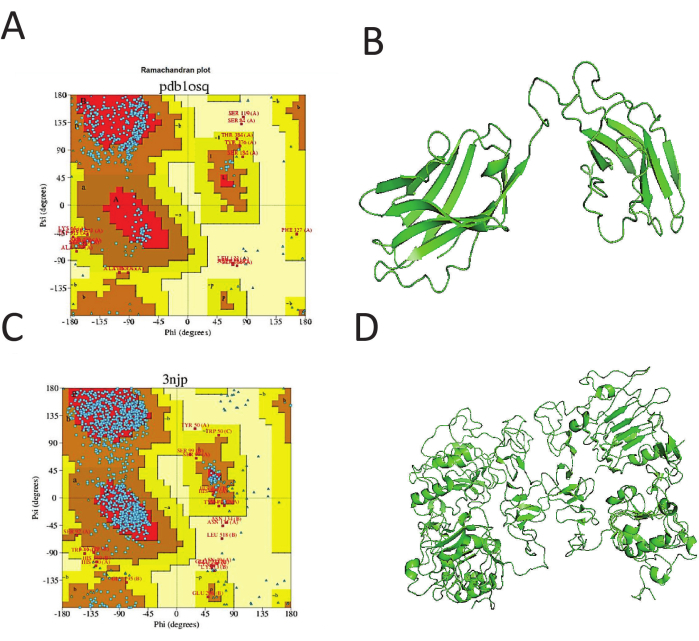
Figure 1: Plot for template selection. The plot is used to determine whether the backbone conformation is correctly predicted. As shown in (A) and (B), most of the peptides are in the correct center of the plot, demonstrating the accuracy and correctness of the structures in (C) and (D). Please click here to view a larger version of this figure.
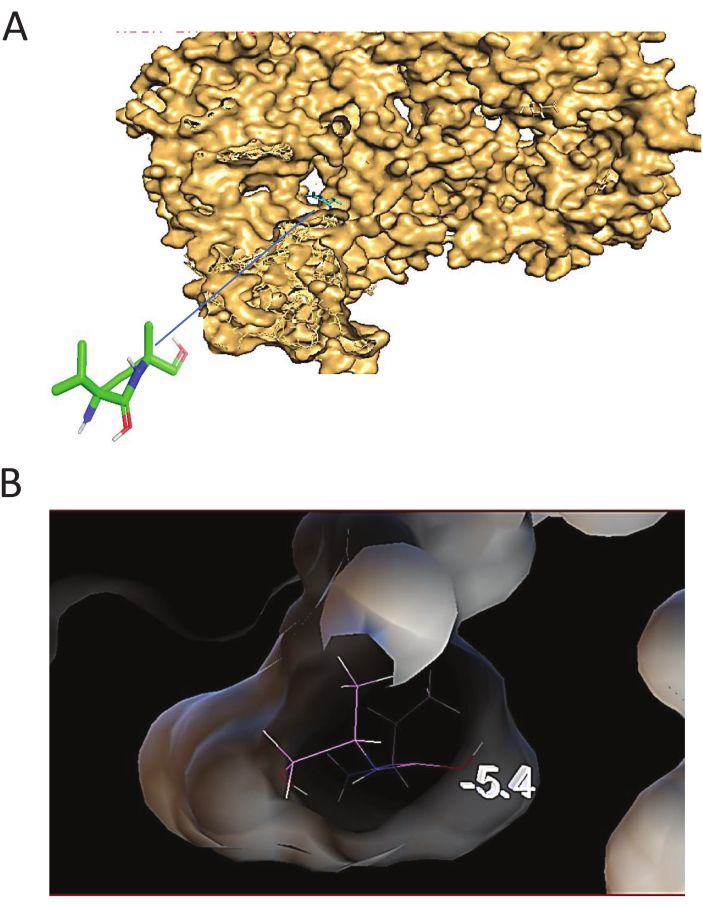
Figure 2: Docking structure and configuration. (A) The Docking structure of the receptor-scFv antibody shows the crystal structure of binding interaction between receptor and ligand in surface structure. (B) Docking configuration. Docking configuration Interaction profile of confirmation and lowest binding presented. Please click here to view a larger version of this figure.
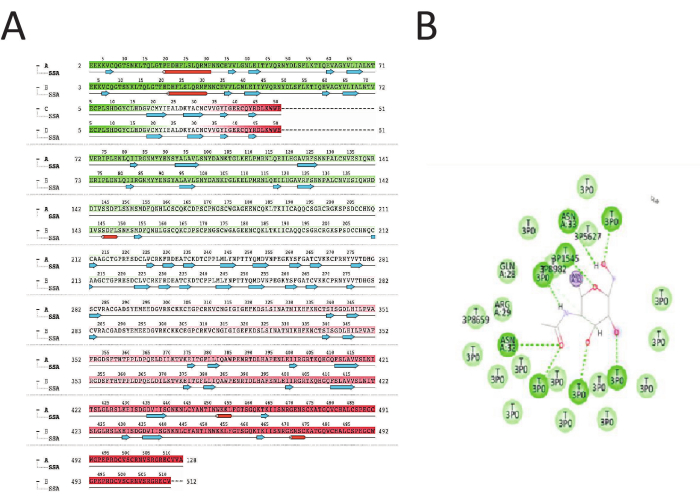
Figure 3: Ligand-receptor residues interaction. (A) Residues interaction of Ligand-receptor structure shows the total number of residue numbers is 1117 residues defined to A =513, B=510, C=7, D =447. A is the receptor. The number of heavy atoms is 8685, while the interaction charges are +55. The dark green alignment is the strongest interaction with the H-bonds. Ligand-receptor residues alignment shows that the green is the strongest bond with the H-bond, the light green is a lighter bond, and the red is the weaker bond. (B) The ligand atoms interact with the protein residues. The Interactions between the ligand and receptor protein occur in more than 30.0% of the simulation time. The receptor residues are connected with the H bond of the ligand atoms. The receptor residues in dark green circles represent the strongest H-bond interaction between the EGFR receptor and scFv antibody. There are eight H bonds, which are considered a strong interaction. Please click here to view a larger version of this figure.
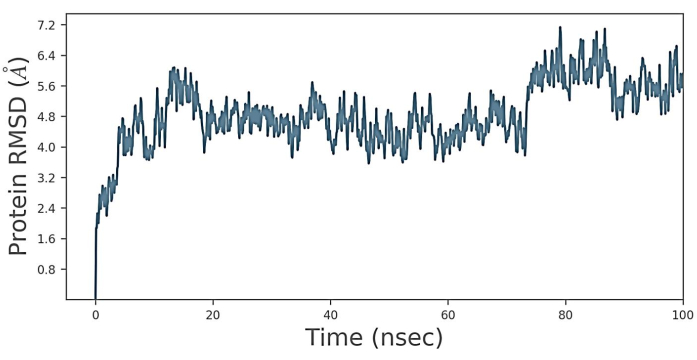
Figure 4: Root mean square deviation (RMSD). RMSD plot of receptor-ligand complex for the 20 ns simulation time. It is observed that protein forms 3.2 Å to 6 Å for the first 0 to 20 ns time step. Additionally, the protein, from 20 ns to 75 ns time step moved. The movement is maintained from 4 Å to 5.6 Å and 75 to 90 ns. The movement increases sharply to 7.2 Å and then decreases to 6.4 A until the last timestep. These results proved that the docked structure EGFR-scFv antibody was stable over the 1000 ns of MD simulation. Please click here to view a larger version of this figure.
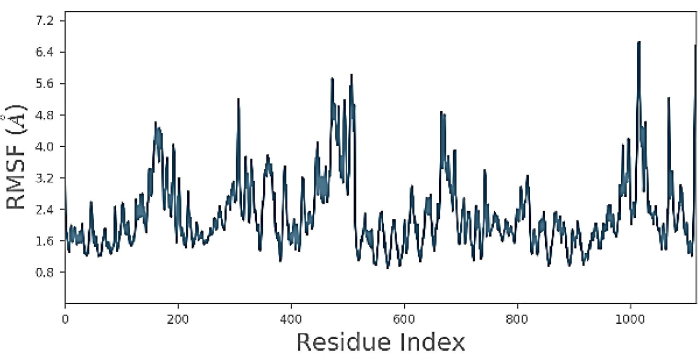
Figure 5: Root mean square fluctuation (RMSF). The figure shows the protein RMSF is moved 2.4 Å from residues between 1-100. then the movement decreases to 2a for 200-300 residues. Further, the protein then fluctuates to 5.6 Å for 500 residues, then sharply declines to 2.4 Å for residues till 1000. There is one sharp peak at the 1000 position with a decrease to 2.4 Å at the terminal. Please click here to view a larger version of this figure.
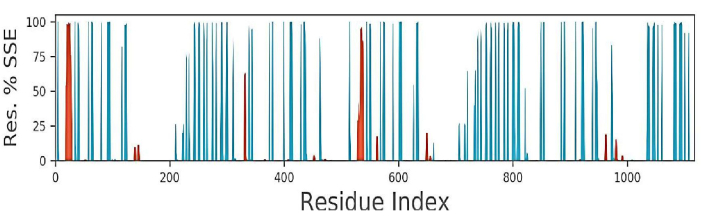
Figure 6: Protein secondary structure elements (SSE) and its residue index. The distribution of secondary structure elements (SSE) by residue index throughout the protein structure is shown in the plot above. The bottom plot tracks each residue's SSE assignment over time, whereas the plot below summarizes the SSE composition for each trajectory frame throughout the simulation. Please click here to view a larger version of this figure.
Figure 7: Simulations of lipid bilayers using the ENERGY force field. Simulations of lipid bilayers using the ENERGY force field. MD simulation is used to investigate geometrical features by showing the cartoon mode. Please click here to view a larger version of this figure.
Supplementary Figure 1: Addition of Kollman charges. Please click here to download this File.
Supplementary Figure 2: Minimizing pdb structure energy. Please click here to download this File.
Supplementary Figure 3: Preparing input files in Python. Please click here to download this File.
Supplementary Figure 4: Steps to create a Ramachandran plot. Please click here to download this File.
Supplementary Figure 5: Ligand creation. Please click here to download this File.
Supplementary Figure 6: Preparation of docking configuration. Please click here to download this File.
Supplementary Figure 7: Molecular dynamic simulation to create the docking complex. Please click here to download this File.
Supplementary Coding File 1. Please click here to download this File.
Supplementary Coding File 2. Please click here to download this File.
Supplementary Coding File 3. Please click here to download this File.
Supplementary Coding File 4. Please click here to download this File.
| タイプ | Total Num. | Concentration [mM] | Charge |
| Cl | 166 | 75.746 | -166 |
| Na | 111 | 50.65 | 111 |
Table 1: Counter ion/salt information.
