In this study, non-woven fiber mats consisting of poly(styrene-butadiene-styrene) fibers in the nano- and micro-scale, were synthesized with and without the presence of iron oxide NPs. To form fibers, the SBS parameters must be carefully selected for the polymer/solvent system used. The molar mass of the dissolved polymer and the solution concentration are critical in controlling the morphology of the structures produced by the SBS process. In this study, a poly(styrene-butadiene-styrene) block-co-polymer (styrene 30 wt. %) was used with a molar mass of approximately 185,000 g/mol and a density of 0.94 g/mL at 25 °C. Multiple studies have examined the effects of polymer molar mass, demonstrating that a higher molar mass favors chain entanglement in solution and drastically increases its viscosity, resulting in fiber formation via the SBS technique21. In addition, previous studies have shown that polymer concentrations in a good solvent (as defined by Flory22) well above the critical overlap concentration (c >> c*), also known as the entanglement concentration (Ce ~ 10c*), will result in fiber formation with minor or no bead formation21,23.
This phenomenon is again governed by the interactions between the entangled polymer chains in solution. Entanglement of the molecules in solution above the c* exponentially increases the viscosity of the solution, therefore overcoming the inertial capillary forces and suppressing the breakup of the polymer jet. Destabilization of the polymer jet after shearing off the polymer solution stream from the nozzle will lead to undesirable "bead" formation if the selected concentration for the SBS experiment is too low. In this study, the critical overlap concentration of the poly(styrene-butadiene-styrene) block-co-polymer in THF was first estimated using the following equation for random polymer coils in a good solvent24:
c* ≈ 3Mw/(4πNARg3) (1)
In equation (1) above, NA, Mw, and Rg are the Avogadro's number, the molar mass of the polymer, and the radius of gyration of the polymer, respectively. This equation estimated the c* of the polymer in solution to be ~8.96 mg/mL. Eight polymer solutions were prepared with different concentrations and their viscosity was studied as a function of concentration. For most polymers, the behavior of their solution's viscosity in a good solvent is linear only at low concentrations.
As polymer concentration increases, the viscosity rises exponentially, and the critical overlap concentration corresponds to the value at which the dissolved polymer coils start to overlap each other and cause entanglement. At that critical concentration, a polymer solution transitions from a dilute to a semi-dilute regime25. The results of the polymer solution's viscosity as a function concentration are shown in Figure 2, and the value of the experimentally estimated c* is ~9.28 mg/mL. The calculated and experimentally predicted values of c* are similar, which is ~10 mg/mL. Therefore, polymer concentration values greater than 10c* (c ≥ 100 mg/mL) were selected to use for the SBS process, to be in the entanglement concentration regime23. At these higher concentrations, the SBS apparatus is capable of consistently producing non-woven fibers with desired diameters and morphology. Figure 3 shows the structure of the developed fiber mats and the morphology of the fibers at a polymer concentration of ~200 mg/mL, N2 gas pressure of approximately 207 kPa, a working distance of nominally 8 cm, and a polymer solution injection rate of ~0.5 mL/min.
The electron micrograph in Figure 3A shows the morphology of the non-woven fiber mat at low magnification. The fiber mat sample consists of primarily individual and cylindrically shaped fibers with minimal polymer beads or polymer welding present. At higher magnification (Figure 3B), it is apparent that the fibers formed are smooth and round, with very similar diameters in the nano scale (diameter range from 100 nm to 600 nm). Individual fibers, as well as some bundle of fibers consisting of 2, 3, and sometimes 4 individual fibers are observed. Finally, the higher magnification images confirm the absence of polymer beads ("beads-on-a-string") or polymer welding under these SBS conditions. To better understand this specific polymer/solvent system and the effect of polymer concentration on the fiber mats produced, the structure and morphology of fiber mat samples sprayed at various concentrations were examined. Significant differences in the fiber mats produced were observed as the polymer concentration increased from approximately 100 mg/mL to 120 mg/mL, 150 mg/mL, and 200 mg/mL, respectively, as seen in Figure 4. The SEM micrographs show a clear transition from fibers that exhibit the undesired "beads-on-a-string" morphology with numerous bundles of fibers present at lower concentrations, near the critical overlap concentration (c* ~10 mg/mL), to the formation of pristine and morphologically smooth fibers at concentrations well above c* (e.g., 200 mg/mL).
Furthermore, as mentioned previously, the gas pressure is another process variable that can influence the morphology and diameter of the produced fibers, although to a much lesser extent than polymer molar mass and concentration. Figure 5 demonstrates the effects of gas pressure, indicating the presence of fibers with decreasing diameter as gas pressure increased from ~138 kPa to ~345 kPa; however, the presence of large polymer beads and welded fibers also increased. Prior work has also demonstrated that very high gas pressures will induce undesirable fiber and polymer welding17,19. This effect could be a result of a more significant decrease in temperature at the spray nozzle when higher gas flow rates are used, due to Joule expansion of the gas. The temperature decrease is proportional to the volumetric expansion of the gas, which in turn, can cause poor solvent evaporation and fiber welding17,19,26. In the current study, based on various SBS parameters and SEM imaging, the optimal polymer concentration and carrier gas pressure for the polymer/solvent system were determined to be 200 mg/mL and 207 kPa.
This combination can consistently produce pristine, smooth, individual fibers in the nano scale (diameter of ~100 nm to 600 nm) without the presence of beads or fiber welding, as shown in Figure 3. It is useful to note that the nitrogen gas was fed to the SBS sprayer through a PTFE tube with an inner diameter of 0.238 cm and length of 2.134 m. At the optimal nitrogen pressure of 207 kPa and approximately 20 °C, the N2 gas density is 0.00215 kg/L, its dynamic viscosity is 1.76 x 10-5 Pa·s, and its approximate velocity is 0.871 m/s with a Reynold's number of 147, indicating a laminar flow. After identifying the best conditions for the SBS parameters in this spray setup for poly(styrene-butadiene-styrene) in THF, the capability of the technique to produce polymer elastomer nanocomposite fiber mats was investigated by dispensing iron oxide NPs in the polymer solution at a mass fraction of χnp = 0.001. This mass fraction was determined to be the highest attainable before destabilization of the NP dispersion was observed. As the NP dispersions were not stable above χnp = 0.001, no dispersions were sprayed at NP mass fractions above this value. Nanoparticle agglomeration phenomena are to be expected, which can later affect the quality of the fibers produced (irregular fiber morphology and diameters) and result in a non-uniform dispersion of the NPs within the fiber material.
It is important to note that after sonication, the iron oxide NP/polymer dispersions at mass fractions equal to 0.001 were stable for approximately 2 hours; therefore, it is recommended to use them immediately after mixing for optimal results. If the dispersions are left unmixed for more than a few hours, it is recommended to sonicate the samples again before beginning SBS. The NPs used in this study, in the form of dry powder, were coated by the manufacturer with silicon oil, which renders them easily dispersible in various organic solvents, including THF. The fiber mats produced were evaluated using backscattered electron (BSE) analysis and EDS in an SEM, and the results demonstrate the presence of iron oxide NPs within the polymer fibers. A representative electron micrograph collected via a BSE detector is shown in Figure 6A. The iron oxide particles (circled in red) can be easily identified in the fibers due to their brighter contrast from the surrounding polymeric fiber material using a BSE detector, as iron is a much heavier element than carbon. In Figure 6C, EDS elemental analysis of the same sample indicates the presence of iron (marked red) at the brighter contrast locations where the iron oxide NPs reside, further validating their presence within the fibers. It is worth noting that the structure and morphology of the fiber mats were not significantly affected by the presence of the iron oxide NPs.

Figure 1: The solution blow spinning apparatus. (A) The apparatus comprises a syringe pump system, an airbrush setup, a collector, an aluminum enclosure, and a nitrogen gas cylinder (not shown); details on the (B) airbrush setup and (C) substrate holder are shown. Please click here to view a larger version of this figure.
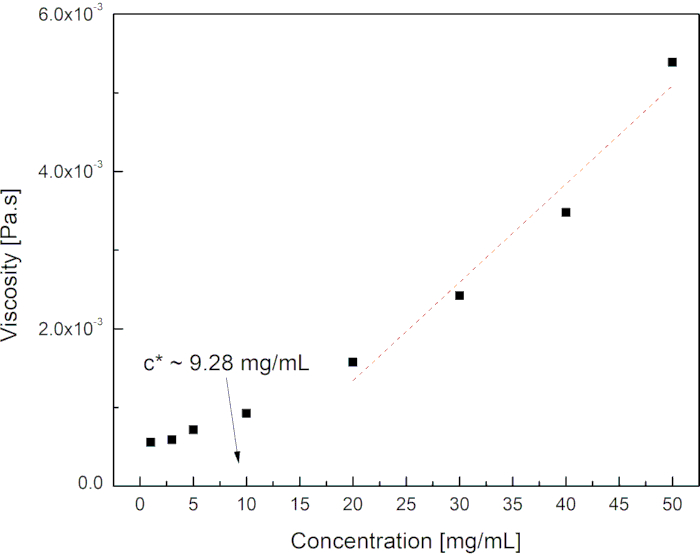
Figure 2: Viscosity of polymer solutions as a function of polymer concentration. The critical overlap concentration (c*) is estimated by the onset of the power law behavior of the viscosity as indicated by the arrow in the graph. Please click here to view a larger version of this figure.

Figure 3: Scanning electron microscopy (SEM) images of poly(styrene-butadiene-styrene) fibers formed via the solution blow spinning (SBS) apparatus. (A) Low-magnification image, and (B) higher-magnification image of the same sample. Scale bar for A = 1 mm; scale bar for B = 40 µm. Please click here to view a larger version of this figure.

Figure 4: SEM micrographs of poly(styrene-butadiene-styrene) solutions sprayed using the SBS apparatus at increasing polymer concentration in solution. Polymer concentration increases from left to right. Scale bars = 40 µm. Abbreviations: SEM = scanning electron microscopy; SBS = solution blow spinning. Please click here to view a larger version of this figure.
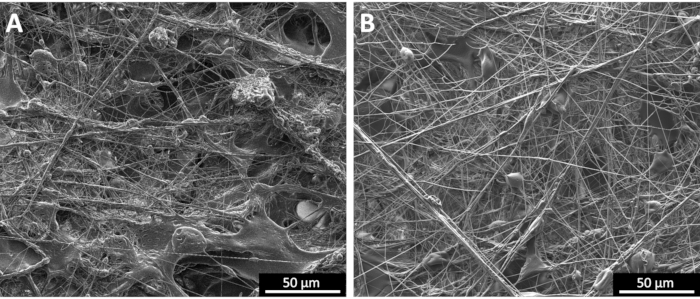
Figure 5: SEM micrographs of poly(styrene-butadiene-styrene) solutions sprayed using the SBS apparatus. (A) High gas pressure of 345 kPa and (B) low gas pressure of 138 KPa. Scale bars = 50 µm. Please click here to view a larger version of this figure.

Figure 6: Backscattered electron micrograph of solution blow spun poly(styrene-butadiene-styrene) fibers. (A) Infused iron oxide (Fe3O4) nanoparticles circled in red; scale bar = 10 µm. (B) Enlargement of the yellow highlighted area at the same magnification. (C) Energy-dispersive X-ray spectroscopy of the enlarged area, indicating the presence of iron (elemental analysis; Fe stained red) within the fibers. Scale bars (B,C) = 4 µm. Please click here to view a larger version of this figure.
