The cell cycle phases of cells from spleen, LNs, and BM of Balb/c mice were analyzed using the fluorescent DNA dye, Hoechst, and an anti-Ki67 mAb, according to the protocol summarized in Figure 1. This staining allowed the differentiation of cells in the following phases of cell cycle: G0 (Ki67neg, with 2N of DNA defined as DNAlow), G1 (Ki67pos, DNAlow), and S-G2/M (Ki67pos, with a DNA content comprised between 2N and 4N, or equal to 4N of DNA defined as DNAintermediate/high).
We first performed cell cycle analysis of BM cells to reproduce previously published results13,14 and then analyzed the cells of interest, i.e., CD8 T cells. Figure 2 shows a typical example of cell cycle analysis of BM cells (Figure 2A). The protocol yielded a low coefficient of variation (CV) of G0/G1 and G2/M DNA peaks, indicating the excellent quality of the DNA staining (Figure 2B, showing an example with CV < 2.5; CV was always < 5 in all the experiments).
We then applied the same protocol to antigen-specific CD8 T cells from vaccinated mice. BALB/c mice were vaccinated against the antigen gag of HIV-1 by using Chad3-gag for priming and MVA-gag for boosting, both engineered to carry HIV-1 gag. At day (d) 3 post-boost, we analyzed the frequency of gag-specific CD8 T cells from the spleen and draining LNs. We took advantage of the newly defined gating strategy for T cells in the early phase of immune response, which in contrast to the conventional strategy, is appropriate for detecting highly activated antigen-responding CD8 T cells12. We executed the novel strategy in five subsequent steps. In step 1, we excluded doublets or aggregates by DNA-A/ -W gate, and in step 2, we identified live cells by dead cell marker exclusion. In step 3, we identified the population of interest using a non-conventional "relaxed" FSC-A/ SSC-A gate (Figure 3A) instead of the canonical narrow lymphocyte gate12. After gating on CD3+CD8+ cells (step 4 of Figure 3A), we identified gag-specific CD8 T cells by using two different MHC multimers, i.e., Pent-gag and Tetr-gag (step 5 of Figure 3A). We used two multimers instead of one to improve the sensitivity of gag-specific CD8 T cell detection in vaccinated mice, without increasing the staining background in untreated mice (Figure 3B and C, step 5). Thus, we successfully distinguished untreated mice (0.00% and 0.00% antigen-specific CD8 T cells in LNs and spleen, respectively) from vaccinated mice (0.46% and 0.29% antigen-specific CD8 T cells in LNs and spleen, respectively, Figure 3B and C).
Notably, the protocol allowed us to have an extremely low background in the antigen-specific CD8 T cell gate of LNs and spleen of untreated mice (usually 0.00% and at maximum 0.02%). The comparison of gag-specific and not gag-specific FSC-A / SSC-A plots showed that the gag-specific cells had high SSC-A and FSC-A (Figure 3D), confirming the need to use a "relaxed" FSC-A/ SSC-A gate to capture these cells. We then evaluated the percentages of gag-specific CD8 T cells in different cell cycle phases (Figure 4A). We found that gag-specific CD8 T cells in the spleen and even more in the draining LNs contained a high proportion of cells in S-G2/M phases at day 3 post-boost (18.60% and 33.52%, respectively).
Furthermore, we found that gag-specific CD8 T cells in S-G2/M phases had high FSC-A and SSC-A, when overlaid onto the total CD8 T cells from the same organ (Figure 4B). CD62L expression by gag-specific CD8 T cells was low, as expected for activated T cells, except for a few cells in G0 in the LNs (Figure 4C). Altogether, these results confirmed that the "relaxed" gate (step 3 of Figure 3A, B, and C) was required to include all of the proliferating antigen-specific CD8 T cells12. The protocol was extremely valuable for a "snapshot" evaluation of cell cycle phases of antigen-specific CD8 T cells at the time of analysis and of CD62L expression by cells in different cell cycle phases.
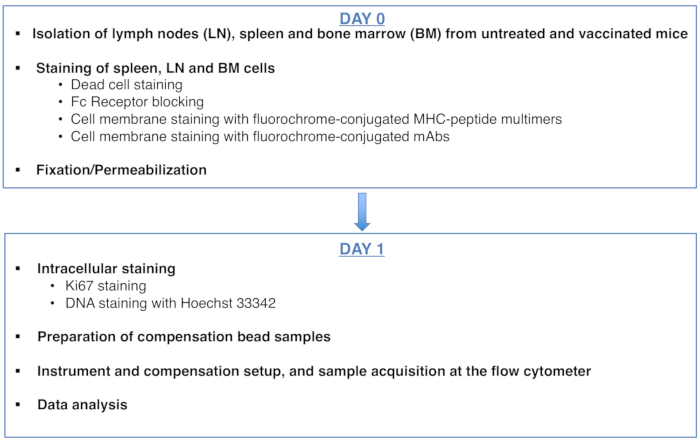
Figure 1: Scheme of the protocol for cell cycle analysis of antigen-specific CD8 T cells. Please click here to view a larger version of this figure.

Figure 2: Cell cycle analysis of BM cells. BM cells from untreated Balb/c mice were stained and analyzed by flow cytometry. (A) Example of gating strategy. We gated on single cells in the DNA-A/-W plot (left) and subsequently on live cells by dead cell dye exclusion (middle). Then, a "relaxed" FSC-A/SSC-A gate was used for all BM cells (right). (B) Example of cell cycle analysis of BM cells (left). We used a combination of Ki67 and DNA staining to identify cells in the following phases of cell cycle: G0 (bottom left quadrant, Ki67neg-DNAlow cells), G1 (top left quadrant, Ki67pos-DNAlow), S-G2/M (top right quadrant, Ki67pos-DNAintermediate/high). Fluorescence Minus One (FMO) control of Ki67 mAb (middle) and DNA histogram (right) are shown. In the DNA histogram plot, the left and right gates correspond to the G0/G1 and the G2/M DNA peak, respectively, and the numbers represent the coefficients of variation (CV) of each peak. In all the other plots, the numbers represent cell percentages in the indicated gates. The figure shows 1 representative experiment out of 5. In each experiment, we analyzed pooled BM cells from 3 mice. Please click here to view a larger version of this figure.

Figure 3: Analysis of antigen-specific CD8 T cells from LNs and spleen. Balb/c mice were primed intramuscularly (i.m.) with Chad3-gag and boosted i.m. with MVA-gag. At day 3 post-boost, draining LN and spleen cells from vaccinated and untreated control mice were stained and analyzed by flow cytometry. (A) Scheme of the gating strategy in five steps to identify single cells (Step 1); live cells (Step 2); lymphocytes (Step 3); CD8 T cells (Step 4); and gag-specific cells (Step 5). (B–C) Example of plots: analysis of cells from (B) LNs and (C) spleen of untreated (top) and vaccinated (bottom) mice. We identified single cells on the DNA-A/ -W plot in Step 1. Then, in Step 2, we selected live cells by dead cell dye exclusion. In Step 3, we used a non-canonical "relaxed" gate for lymphocytes. In Step 4, we identified CD8 T cells by their double expression of CD3 and CD8. We then identified gag-specific cells and not gag-specific in Step 5, based on their capacity to bind fluorochrome-labelled H-2kd-gag-Pentamer (Pent-gag) and H-2kd-gag-Tetramer (Tetr-gag), or not, respectively. (D) FSC-A/SSC-A profiles of gag-specific (blue) and not gag-specific (grey) cells after gating as described above. Numbers represent cell percentages in the indicated gates. The figure shows 1 representative experiment out of 5. In each experiment, we analyzed pooled spleen and pooled LN cells from 3 vaccinated mice and 3 untreated mice. Please click here to view a larger version of this figure.

Figure 4: Cell cycle analysis of antigen-specific CD8 T cells. Mice were vaccinated as in Figure 3 and cell cycle analysis of gag-specific cells was performed at day 3 post-boost, after gating in 5 steps as in Figure 3. (A) Example of cell cycle analysis of gag-specific CD8 T cells from LNs (top) and spleen (bottom) of vaccinated mice. Cell cycle phases were identified as in Figure 2B. The panels represent cells in G0, in G1, and in S-G2/M (left) and Fluorescence Minus One (FMO) control of Ki67 mAb (right). Numbers represent cell percentages in the indicated gates. (B) FSC-A/SSC-A dot plots showing gag-specific CD8 T cells in S-G2/M phases (in red) overlaid onto total CD3+CD8+ T cells (in grey) from LNs (top) and spleen (bottom) of vaccinated mice. (C) Offset histograms showing CD62L expression by gag-specific CD8 T cells in G0 (green), in G1 (blue), and in S-G2/M (red) from LNs (top) and spleen (bottom) of vaccinated mice. The y-axes indicate normalized number of events. The figure shows 1 representative example out of 5 independent experiments with a total of 15 mice. Please click here to view a larger version of this figure.
Supplementary Material: Flow cytometer settings. Please click here to download this file.
