Preparation of Meiotic Chromosome Spreads from Zebrafish Spermatocytes
概要
Nuclear surface spreads are an indispensable tool for studying chromosome events during meiosis. Here we demonstrate a method to prepare and visualize meiotic chromosomes during prophase I from zebrafish spermatocytes.
Abstract
Meiosis is the key cellular process required to create haploid gametes for sexual reproduction. Model organisms have been instrumental in understanding the chromosome events that take place during meiotic prophase, including the pairing, synapsis, and recombination events that ensure proper chromosome segregation. While the mouse has been an important model for understanding the molecular mechanisms underlying these processes, not all meiotic events in this system are analogous to human meiosis. We recently demonstrated the exciting potential of the zebrafish as a model of human spermatogenesis. Here we describe, in detail, our methods to visualize meiotic chromosomes and associated proteins in chromosome spread preparations. These preparations have the advantage of allowing high resolution analysis of chromosome structures. First, we describe the procedure for dissecting testes from adult zebrafish, followed by cell dissociation, lysis, and spreading of the chromosomes. Next, we describe the procedure for detecting the localization of meiotic chromosome proteins, by immunofluorescence detection, and nucleic acid sequences, by fluorescence in situ hybridization (FISH). These techniques comprise a useful set of tools for the cytological analysis of meiotic chromatin architecture in the zebrafish system. Researchers in the zebrafish community should be able to quickly master these techniques and incorporate them into their standard analyses of reproductive function.
Introduction
Sexual reproduction proceeds through the combination of two haploid gametes, each carrying half the chromosome complement of a somatic cell. Meiosis is a specialized cell division that produces haploid gametes through one round of DNA replication and two successive rounds of chromosome segregation. In prophase I, homologous chromosomes (homologs) must undergo pairing, recombination, and synapsis, the latter of which is characterized by the formation of the synaptonemal complex that comprises two homolog axes bridged by the transverse filament, Sycp1 (Figure 1A,B). Failure to properly execute these processes can lead to the production of aneuploid gametes, which are a leading cause of miscarriages in humans1. Our knowledge of the coordination between pairing, recombination and synapsis has been facilitated by studies in a wide range of organisms, such as yeast, C. elegans, mouse, and Drosophila, among others2. While the general process of homologous chromosome pairing followed by segregation is well conserved, its dependency on recombination and synapsis and the order of these events varies.
Meiotic double-strand break (DSB) formation, which initiates homologous recombination, occurs near telomeres clustered in the bouquet during leptotene and synapsis ensues shortly after3,4. This configuration of DSB formation and synapsis initiation is also a characteristic of male meiosis in humans but not in mouse5,6,7,8, suggesting that zebrafish can serve as a model for human spermatogenesis. There are also several practical advantages of studying zebrafish meiosis. Both males and females undergo gametogenesis throughout adulthood, their gonads are easily accessible, and hundreds of offspring are generated from a single cross. Additionally, the embryos are transparent and develop externally, which facilitates the early detection of aberrations in embryonic development due to aneuploid gametes3,9. Disadvantages of using zebrafish are that they are slow to reach sexual maturity (~60 days) and the amount of material needed for nuclear surface spreads must be collected from ~10-20 adult animals, depending on their size.
Meiotic chromosome spread preparations are a vital tool for studying chromosome dynamics across all model organisms, since key signatures of meiotic chromosome dynamics can be probed. In zebrafish, key aspects of the progression of the meiotic program and nuclear organization have been dissected through probing nuclear surface spreads, referred to here as chromosome spreads, with antibodies for immunofluorescence detection of proteins and/or nucleic acids by FISH3,4,9,10,11,12. Indeed, the polarized localization of clustered telomeres in the bouquet can be preserved in the spread preparation (Figure 1C). Recently, we have used zebrafish spermatocyte chromosome spreads together with fluorescence detection methods and super-resolution microscopy to elucidate the detailed progression of zebrafish telomere dynamics, homologous chromosome pairing, double-strand break localization, and synapsis at key meiotic transitions3. Here we present methods to prepare chromosome spreads from spermatocytes of the zebrafish testes and subsequently stain them with fluorescent peptide nucleic acid (PNA) probes to repeated telomere sequences and immunofluorescence detection of chromosome associated proteins.
Protocol
All methods involving zebrafish were carried out using ethical standards approved by the Institutional Animal Care and Use Committee at UC Davis.
1. Chromosome spreading procedure
NOTE: The following protocol is designed to create 4-6 slides, with hundreds of spread meiotic nuclei per slide. The numbers of testes used will depend on the size of the fish. Expect to use 20 animals at ~60 days post fertilization (dpf) and 15 animals at ~6 months post fertilization (mpf). For large zebrafish (e.g., 12 mpf) 10 animals should be sufficient. Be aware that testes of some meiotic mutants (e.g., spo11-/-) will be somewhat smaller3. In this protocol, one pool of testes is considered as a single sample. It is recommended that no more than 4 samples are prepared in parallel.
- Make the following solutions before starting the spreading procedure
NOTE: It is convenient to prepare the following solutions in the quantities specified for use in future spreading experiments.- Prepare a 0.1 M sucrose solution by dissolving 3.42 g of sucrose in 100 mL of distilled water. Bring the sucrose solution to pH 8 using 1 M Tris-HCl that is also at pH 8. Then filter sterilize the sucrose solution. Store at room temperature.
- Make a 10x phosphate-buffered saline (PBS) stock solution to a final concentration of 1.37 M NaCl, 27 mM KCl, 73 mM Na2HPO4 and 27.6 mM KH2PO4 in 1.8 L of distilled water. Stir until dissolved, then adjust pH to 7.3 using NaOH and autoclave. Store at room temperature.
- Make a 400 µg/mL DNase I solution in sterile distilled water. Store at -20 °C. It is recommended to prepare 5 mL of this solution and then store as 100 µL aliquots.
- Make the following solutions on the day of the spreading protocol
NOTE: For time efficiency, it is best to make the collagenase solution immediately after dissecting all testes and then make the trypsin and trypsin inhibitor solutions during the time that the samples are being treated in collagenase.- Dissolve 4 mg of collagenase in 200 µL of Dulbecco's modified Eagle medium (DMEM) per sample (2% w/v collagenase final concentration). Keep on ice until needed. The collagenase will dissociate the testes.
- Dissolve 1.4 mg of trypsin in 200 µL of DMEM per sample (0.7% w/v trypsin final concentration). Keep on ice until needed. The trypsin will help dissociate the cells.
- Dissolve 10 mg of trypsin inhibitor in 500 µL of DMEM per sample (2% w/v trypsin inhibitor final concentration). Keep on ice until needed. Trypsin inhibitor prevents trypsin from degrading the cells.
- Prepare a 1% formaldehyde solution (from a 16% pre-made solution) with 0.15% Triton X-100 in sterile distilled water. Keep on ice until needed. The 1% formaldehyde can be prepared while the testes are being treated with trypsin and DNase I (i.e., during step 1.4.7 of the spreading procedure).
CAUTION: Formaldehyde is hazardous.
- Dissection of testes from adult male zebrafish
- Euthanize male zebrafish (> 60 dpf) by submerging them in ice water. The fish can be kept in ice water until ready to dissect, however they should be dissected as soon as possible.
NOTE: The wild-type strain AB is used for this procedure, but other wild-type strains should also be amenable. The longest time we have kept fish in ice water prior to dissecting is 3 h without any noticeable effect on the protocol. - Decapitate one fish at a time with small scissors and then use micro scissors to cut along the ventral midline to expose the body cavity.
- Dissect the testis using forceps (see Table of Materials) at 1.65x magnification under a microscope. Carry out the dissections in a silicone-coated Petri dish with covered by a shallow pool of 1x PBS.
NOTE: The testis is located in between the swim bladder and the intestine and will appear lighter than muscle tissue (Figure 2A). The testis should have 2 lobes when removed from the zebrafish (Figure 2B). - Remove as much fat and surrounding tissue from the testis as possible. Then add each dissected testis directly into a 5 mL tube with 2 mL of DMEM and keep on ice.
NOTE: Steps 1.3.3 and 1.3.4 should be mastered prior to doing any experiments.
- Euthanize male zebrafish (> 60 dpf) by submerging them in ice water. The fish can be kept in ice water until ready to dissect, however they should be dissected as soon as possible.
- Dissociation of testes cells
- Pre-warm 100 mL of 0.1 M sucrose solution to 37 °C.
- Add 200 µL of collagenase solution to the 5 mL tube with the testes. Mix the solution by inverting it several times.
- Gently shake the testes in an incubator shaker horizontally at 100 rpm at 32 °C for 50 min to an hour until the DMEM is cloudy and the testes are in small chunks. Rapidly invert the tube every 10 min to facilitate dissociation.
- To wash out the collagenase, add DMEM to a final 5 mL volume and invert the tube a few times. Pellet the testes at ~200 x g for 3 min at room temperature. Remove 3 mL of the supernatant so that only 2 mL remains.
NOTE: The addition, removal, and transfer of DMEM is done with plastic transfer pipettes for the entirety of the chromosome spread procedure. - Repeat step 1.4.4 twice more for a total of 3 DMEM washes. Do not resuspend the pellet between DMEM washes. After the last DMEM wash, remove 4 mL of the supernatant so that only 1 mL remains.
- Add 1 mL of DMEM for a total volume of 2 mL and add 200 µL of the trypsin solution and 20 µL of DNase I solution. Invert the tube a few times to mix the solution.
- Horizontally shake the tube at 32 °C for 5-15 min at 100 rpm until the DMEM solution contains only a few clumps. Rapidly invert the tube every 5 min to facilitate dissociation.
NOTE: The clumps should be considerably smaller after shaking. - Add 500 µL of the trypsin inhibitor solution and 50 µL of DNase I solution. Invert the tube a few times to mix, then briefly spin down at ~200 x g for 3 min at room temperature to remove liquid or clumps that may adhere to the tube cap or the side of the tube.
- Place the tube on ice and resuspend the cell suspension by repeatedly pipetting up and down with a plastic transfer pipette for 2 min to facilitate dissociation of any remaining clumps. Do not let the cell suspension go into the bulb of the transfer pipette. After the 2 min, pipette the suspension back into the 5 mL tube.
- Pre-wet a 100 µm cell strainer with DMEM and place it on top of a 50 mL tube on ice.
- Transfer the cell suspension through the strainer one drop at a time using a plastic transfer pipette. Ensure that the cell suspension does not go into the bulb of the transfer pipette.
- Transfer the filtrate using a plastic transfer pipette to a new 5 mL tube and add DMEM to a final volume of 5 mL. Be sure to collect the pooled cells attached to the underside of the filter with a clean plastic transfer pipette. Pellet the cells at ~200 x g for 5 min at room temperature.
- Remove as much supernatant as possible without disturbing the pellet.
- Add 5 µL of DNase I solution directly into the pellet. Then mix the pellet with the DNase I solution by gently scraping the outside bottom of the tube 4-5 times along an empty 1.5 mL microcentrifuge tube rack. Scraping too harshly can result in a reduction of recovered spreads.
- Add DMEM to a final volume of 5 mL and mix the solution by inverting the tube several times. It is common for clumps to still be present after the first DNase I treatment.
- Pellet the cell suspension at ~200 x g for 2 min at room temperature.
- Repeat steps 1.4.13-1.4.16 an additional 1-3 times until the resuspended pellet does not clump upon addition of DMEM. After the last spin, remove as much supernatant as possible without disturbing the pellet.
NOTE: Expect a reduction in pellet size with every DNase I treatment; exceeding 4 total DNase I washes may result in significant loss of cells. If no pellet is visible after the DNase I treatments steps, do not proceed with the procedure. Discard any unused DNase I. - Add 1 mL of 1x PBS and resuspend the pellet by gently scraping the outside bottom of the tube along an empty 1.5 mL tube rack.
- Pellet the cell suspension at ~200 x g for 5 min then remove as much supernatant as possible without disturbing the pellet.
- Cut ~3 mm from the end of a 200 µL pipette tip to widen the aperture and resuspend the pellet in ~80 µL of 0.1 M sucrose solution by pipetting up and down with the cut pipette tip. Let the cell suspension sit at room temperature for 3 min.
- Spreading chromosomes on glass slides
- Coat one slide with 100 µL of 1% formaldehyde with 0.15% Triton X-100 with the side of a pipette tip. Then add 18 µL of the sucrose cell suspension onto the center of the slide in a straight line perpendicular to the long edge. Tilt the slide back and forth (~60°) to facilitate spreading of the cell suspension to all corners.
- Place the slides flat down in a slightly cracked open humidity chamber (Figure 3) to prevent the formaldehyde solution from drying. Place the humidity chamber in a dark drawer overnight.
- Remove the lid from the humidity chamber and allow the slides to fully dry.
- Place the slides in a Coplin jar then fill the Coplin jar with distilled water and incubate for 5 min with gentle shaking at room temperature.
NOTE: The slides can be placed in the Coplin jar in a zigzag pattern to maximize the number of slides per Coplin jar. Make sure there is space for liquid to pass between all the slides. - Pour out the water and fill the jar with 1:250 wetting agent (see Table of Materials). Wash 2 times for 5 min each with gentle shaking at room temperature.
- Allow the slides to fully dry and store at -20 °C until they are stained.
NOTE: The longest we have stored slides without any noticeable degradation of the chromosomes is ~2 months.
2. Telomere PNA probe staining
NOTE: Telomere repeats can be stained using fluorophore-conjugated telomere PNA probes that hybridize to leading strand telomere repeats (CCCTAA). PNA probes have a neutral backbone, which increases hybridization affinity to negatively charged DNA, resulting in little to no background. The telomere probing step is optional. To proceed to antibody staining, rehydrate slides in 1x PBS as indicated in step 2.2.2., then proceed directly to "Primary antibody staining".
- Make the PNA probe reagents before starting the telomere staining
NOTE: It is convenient to prepare the following solutions in the quantities specified for use in future experiments. Storage conditions are noted below.- Prepare 2 L of 20x saline-sodium citrate (SSC) solution with a final concentration of 3 M NaCl and 0.3 M sodium citrate. Autoclave and store at room temperature.
- Make 50 mL of pre-hybridization solution containing transfer RNA to a final concentration of 50% formamide, 5x SSC, 50 µg/mL heparin, 500 µg/mL transfer RNA, 0.1% Tween 20 and add 460 µL of 1 M citric acid to bring the solution to pH ~6. Store at -20 °C.
CAUTION: Formamide is hazardous. Prepare the solution in a fume hood. - Make 50 mL of pre-hybridization solution prepared in the same way as in step 2.1.2 without adding transfer RNA. Store at -20 °C.
- PNA telomere probes TelC-Cy3 and TelC-Alexa647 are prepared as 50 µM stocks in formamide as per manufacturer's instructions. Store the PNA probes as 8 µL aliquots at -80 °C.
- Make at least 1 mL of 100 mg/mL stock solution of bovine serum albumin (BSA) in sterile distilled water. Store at -20 °C. If preparing more than 1 mL of stock BSA, store the solution as 1 mL aliquots.
- Using a 2 mL tube, prepare 2 mL of hybridization solution by adding 27 µL of 100 mg/mL BSA and 8 µL of 50 µM PNA probe to 1.965 mL of pre-hybridization solution containing transfer RNA. Store in the dark at -20 °C.
- PNA telomere probe staining
- Preheat the hybridization oven to 82 °C.
- Re-hydrate slides with 500 µL of 1x PBS per slide for 5 min in a humidity chamber at room temperature. Remove the PBS by tapping the side of the slide on a paper towel.
- Heat the slides and the 2 mL tube containing the hybridization solution directly on a metal surface in the heated hybridization oven for 2 min.
- While keeping the slides on the metal surface, add 100 µL of the hybridization solution per slide and then cover the slides with plastic coverslips (~25 mm x 75 mm) cut to shape out of an autoclave bag. Let slides sit at 82 °C for 10-12 min.
- Place the slides in a humidity chamber in the dark at 37 °C for 16-24 h. From this point forward and for the primary and secondary antibody staining, the slides must be kept in the dark to avoid photobleaching of the fluorescence signal. After the incubation period, the reagents for antibody staining can be prepared during step 2.2.9.
- Remove the coverslips with a pipette tip.
NOTE: To facilitate removal of the coverslip, ~50-100 µL of the hybridization solution with no transfer RNA can be gently dispensed between the slide and the coverslip as the coverslip is being lifted. - Pipette 500 µL of pre-hybridization solution with no transfer RNA onto each slide and place the slides in the humidity chamber for 15 min at room temperature. Remove the solution by tapping the side of the slide on a paper towel.
NOTE: The pre-hybridization solution with no transfer RNA is not pre-heated prior to being added onto each slide. - Pipette 500 µL of 50% pre-hybridization solution with no transfer RNA in 1x PBS to each slide and place the slides in the humidity chamber for 15 min at room temperature. Remove the solution by tapping the side of the slide on a paper towel.
- Transfer the slides to a Coplin jar and wash 3 times in 1x PBS with gentle shaking for 15 min per wash at room temperature.
- Remove the slides from the Coplin jar and remove excess PBS by tapping the side of the slide on a paper towel. Then proceed to step 3.2 "Primary antibody staining".
3. Antibody staining
NOTE: Antibodies raised to known meiotic proteins can be used for immunofluorescence detection in spread chromosome preparations. Secondary antibodies conjugated to different fluorophores allows for multiple proteins to be stained simultaneously, if the primary antibodies were raised in different animals.
- Make the following solutions on the day of the primary and secondary antibody staining
- Make 1 L of PBT with 1x PBS and 0.1% Triton X-100 diluted in distilled water. Store at room temperature. This can be prepared ahead of time and in large quantities for future spreading experiments.
- Prepare 500 µL of antibody block per slide to a final concentration of 2 mg/mL of BSA and 2% goat serum in PBT.
- To make 100 µL of primary or secondary antibody mix per slide, add the appropriate antibodies at the correct concentration (see Table of Materials) into the antibody block.
NOTE: Antibody concentration may need to be determined empirically. In the discussion, we include a list of antibodies we have tried with success (with dilution/concentrations) and those we have tried without success.
- Primary antibody staining
NOTE: Rabbit anti-human SCP3 and chicken anti-zebrafish Sycp1 are typically used for primary antibody staining.- After removing excess PBS from the slides, add 500 µL of antibody block per slide and place the slides in a humidity chamber at room temperature for a minimum of 20 min.
- Remove the antibody block by tapping the side of slide on a paper towel.
- Add 100 µL of primary antibody mix in antibody block per slide and cover the slides with a plastic coverslip made from an autoclave bag. Place the slides in a humidity chamber overnight at 4 °C.
- Remove the coverslips with a pipette tip and wash the slides 2 times for a minimum of 5 minutes each with 1x PBS in a Coplin jar with gentle shaking at room temperature.
- Remove the PBS by tapping the side of the slide on a paper towel.
- Secondary antibody staining
NOTE: Conjugated goat anti-rabbit and anti-chicken antibodies to different fluorophores are used for staining at a 1:1000 concentration.- Add 500 µL of antibody block per slide and place the slides in the humidity chamber at room temperature for a minimum of 5 min.
- Remove the antibody block by tapping the side of the slide on a paper towel.
- Add 100 µL of secondary antibody mix in antibody block per slide and cover the slides with a plastic coverslip made from an autoclave bag. Place the slides in a humidity chamber for 1 h at 37 °C.
- Remove the coverslips with a pipette tip and wash the slides 3 times for a minimum of 5 min each with 1x PBS in a Coplin jar with gentle shaking at room temperature.
- Rinse the slides one time for a minimum of 2 min with distilled water in a Coplin jar with gentle shaking at room temperature.
- Air dry the slides tilted, to facilitate drying.
- Place 20 µL of anti-fade mountant with or without DAPI (see Table of Materials) as 3 evenly spaced dots on glass coverslips (24 mm x 60 mm).
- Place the slides face down on the glass coverslips then seal the coverslips with nail polish on the edge overlapping the frosted end of the slide.
- Store the prepared slides at 4 °C until ready for imaging.
Representative Results
We have outlined a method to prepare and visualize zebrafish spermatocyte spread preparations. When performed correctly, our procedure yields well spread, non-overlapping nuclei. To recover such nuclei, it is important to have the appropriate amount of starting material (i.e., testes), treat testes for a sufficient length of time in trypsin and an adequate number of DNase I treatments. These spreads can then be stained for telomeres and meiotic proteins to study meiotic progression during prophase I. Figure 1 depicts examples of spread preparations stained for chromosomal features at different stages of prophase I. Figure 4 illustrates an example of poorly spread nuclei.
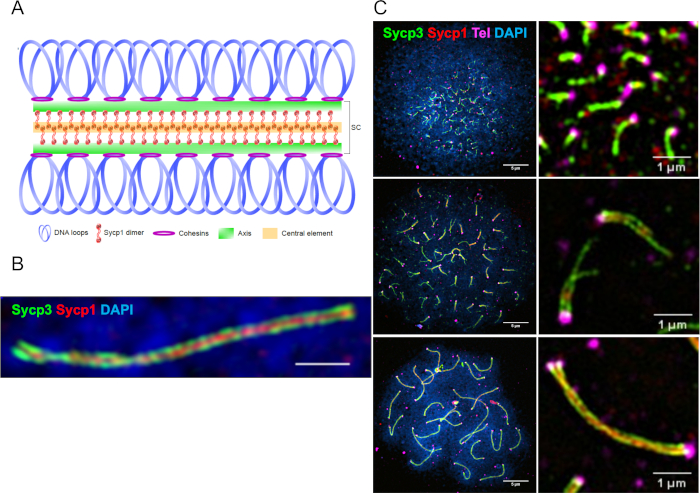
Figure 1: Representative super-resolution images of chromosome spread preparations stained with PNA and antibody probes. (A) Schematic of the synaptonemal complex. (B) A synapsed pair of homologs showing the chromosome axis protein Sycp3 (green), the transverse filament protein Sycp1 (red), and DNA (Blue) imaged by structural illumination microscopy (SIM) using a 100x objective. The scale bar = 1 µm. (C) The panels on the left show three stages of meiotic prophase: leptotene (top), early-zygotene (middle) and pachytene (bottom). The scale bar = 5 µm. Right: representative images containing telomeres (magenta) for each stage. The scale bar = 1 µm. Figures are adapted from Blokhina et al.3. Please click here to view a larger version of this figure.

Figure 2: Example of zebrafish testes (~7 months post fertilization). (A) Image shows the relative location of the testis within the zebrafish. The testis sits between the swim bladder and the intestine. (B) The length of a testis. Younger zebrafish will have smaller testes. For chromosome spreads, remove as much fat and surrounding tissue as possible from the testis. Please click here to view a larger version of this figure.
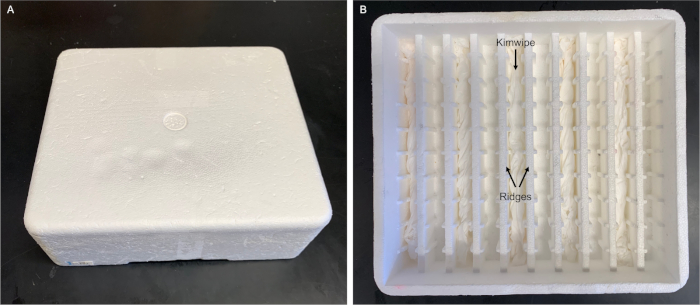
Figure 3: Humidity chamber for slides. Our humidity chamber is made using a polystyrene foam box (21 cm x 19 cm x 6 cm) designed to ship electroporation cuvettes. We inserted wet thin tissue wipes (see Table of Materials) in every other groove. The slides are placed flat on top of the ridges without touching the wet wipes. A commercial humidity chamber is also available (see Table of Materials). We envision that any polystyrene foam box fitted with ridges and grooves (e.g., using glass pipettes13) can be used. Please click here to view a larger version of this figure.
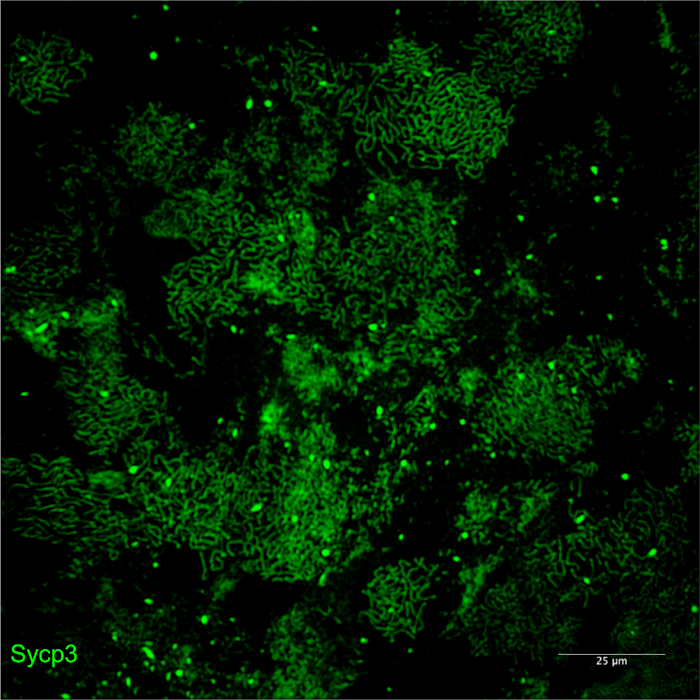
Figure 4: Examples of poor-quality spreads due to insufficient DNase I treatments. Meiotic chromosomes stained for Sycp3 imaged by SIM using a 20x objective. Insufficient DNase I treatments leads to overlapping nuclei due to a viscous sucrose cell suspension that prevents cells from properly spreading on the slide. Please click here to view a larger version of this figure.
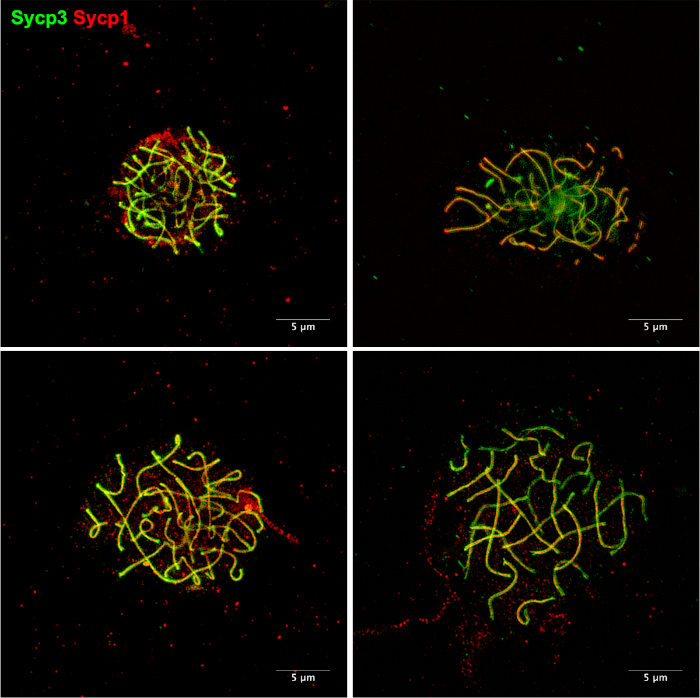
Figure 5: Examples of chromosome spreads using an alternative method11. Meiotic chromosomes stained for Sycp3 (green) and Sycp1 (red). Chromosome spreads were prepared as described by Sansam and Pezza11. This method can be performed using individual zebrafish rather than the several zebrafish required for our protocol. In our hands, it is common to see chromosomes that are not well-spread. There is also a noticeable increase in background staining that results from debris that is left on the slides during the chromosome spreading procedure. Please click here to view a larger version of this figure.
Discussion
Here we describe methods to probe the location of telomeres and chromosome-associated proteins in nuclear surface spreads from spermatocytes isolated from zebrafish testes. We expect that these methods will be applicable for analysis of spermatocytes in other teleost species with adjustment to the size of the testis.
While only a few antibodies have been raised to zebrafish meiotic proteins, we have had success using the following antibodies raised to human (h) or mouse (m) proteins. Our lab has raised antibodies to zebrafish Sycp1 protein (zfSycp1) in chicken that has served as a reliable marker for the synaptonemal complex (SC), however, this protein is present only from early zygotene to late pachytene, when chromosomes are fully synapsed. The rabbit anti-hSYCP3 antibody has been very reliable to detect meiotic chromosome axes from leptotene until the dissolution of the SC in late pachytene (Figure 1B,C). Notably, we have not observed a classical diplotene stage with full-length axes defined by Sycp3 and the absence of Sycp1, suggesting that, unlike in mouse, the axes may be degraded after exit from pachytene3. Other commercially available antibodies to m, h or zf meiotic proteins that have given positive results also include DNA recombination/replication rabbit anti-hRAD51 and rabbit anti-hRPA. The source of each antibody and the dilution used is listed in the Table of Materials. Those we have tried without success using at least three different dilutions include goat anti-hDMC1, mouse anti-hMlh1, mouse anti-hamster Sycp3, mouse anti-hRPA.
There are three critical stages of the chromosome spread procedure. The first is collecting the correct amount of material. Fifteen intact testes should be sufficient for the spreading protocol for ~6 mpf males, and this number can be adjusted up or down depending on the size of the animals. Keep in mind that some meiotic mutants will have smaller testes, so 2 additional animals should be used. The second important step is the dissociation of cells. During trypsin digestion, if the testes do not dissociate into small clumps, it is likely that too few cells will be recovered from the spreading procedure. A third key step is the presence of a visible pellet after the DNase I treatments. If a pellet is not visible, it is likely that the spreading procedure will yield little to no nuclei. Loss of a pellet might arise if too few animals are used or if too many DNase I washes are carried out. On the other hand, too few DNase I treatments can result in a viscid sucrose cell suspension, which will prevent cells from spreading on the slide (Figure 4). It is important to continue performing DNase I treatments (for a maximum of 4 treatments) until clumps are no longer present upon addition of DMEM.
The presented chromosome spreading technique yields hundreds of well spread nuclei per slide. Using this procedure, we have been able to provide a detailed analysis of key events during zebrafish spermatogenesis using super-resolution microscopy3. In our hands, this protocol has generated better spread chromosomes, with less debris on the slide, and less background staining compared with faster methods using single animals described by Moens14 and Sansam and Pezza11 (Figure 5). These differences are likely due to the use of collagenase, trypsin, and DNase treatments in our protocol. Two limitations of our technique are the requirement of using 10-20 adult zebrafish males and the more laborious enzyme treatments and washing steps. If super-resolution detection is not necessary, if the zebrafish material is limited, or more than two conditions are tested in parallel, the faster methods may be more suitable alternatives14,15,16.
開示
The authors have nothing to disclose.
Acknowledgements
We thank Trent Newman and Masuda Sharifi for comments on the manuscript and An Nguyen for helping to optimize methods for spreading and staining chromosomes from zebrafish meiocytes. This work was supported by NIH R01 GM079115 awarded to S.M.B.
Materials
| 1.5 mL centrifuge tubes | Several commercial brands available | ||
| 1.5 mL microcentrifuge tube rack | Several commercial brands available | ||
| 16% formaldehyde, methanol-free | ThermoFisher Scientific | 28908 | |
| 2 mL | Several commercial brands available | ||
| 24 x 50 mm glass coverslips | Corning | 2980-245 | |
| 24 x 60 mm glasscoverslips | VWR International | 16004-312 | |
| 50 mL conical centrifuge tubes | ThermoFisher Scientific | 363696 | |
| Autoclave bag | Several commercial brands available | Used to make plastic coverslips. | |
| Bovine Serum Albumin (BSA) | Fisher Scientific | BP1605-100 | Prepare a 100 mg/ml stock solution in sterile distilled water. |
| Cell Strainer, 100 µm | Fisher Scientific | 08-771-19 | |
| CF405M goat anti-chicken IgY (H+L), highly cross-adsorbed | Biotium | 203775-500uL | Use at 1:1000 |
| Chicken anti-zfSycp1 | Generated by Burgess lab | N/A | Use at 1:100 |
| Collagenase from Clostridium histolyticum | Sigma-Aldrich | C0130-500MG | |
| Coplin jar | Several commercial brands available | ||
| DNase I, grade II from bovine pancreas | Roche Diagnostics | 10104159001 | |
| Dulbecco's Modified Eagle Medium (DMEM) | Fisher Scientific | MT10014CV | |
| Dumont No. 5 Forceps | Fine Science Tools | 11252-30 | Two are required for dissecting the testes. |
| Eppendorf Tubes, 5 mL | VWR International | 89429-308 | |
| Formamide | Fisher Scientific | BP228-100 | |
| Goat anti-chicken IgY (H+L) secondary antibody, Alexa Fluor 488 | ThermoFisher Scientific | A-11039 | Use at 1:1000 |
| Goat anti-chicken IgY (H+L) secondary antibody, Alexa Fluor 594 | ThermoFisher Scientific | A-11042 | Use at 1:1000 |
| Goat anti-hDMC1 | Santa Cruz Biotechnology | sc-8973 | Does not work in our hands |
| Goat anti-rabbit IgG (H+L) cross-adsorbed secondary antibody, Alexa Fluor 488 | ThermoFisher Scientific | A-11008 | Use at 1:1000 |
| Goat anti-rabbit IgG (H+L) cross-adsorbed secondary antibody, Alexa Fluor 594 | ThermoFisher Scientific | A-11012 | Use at 1:1000 |
| Goat serum | Sigma-Aldrich | G9023-10mL | |
| Heparin sodium salt | Sigma-Aldrich | H3393-100KU | |
| Humidity chamber | Fisher Scientific | 50-112-3683 | |
| Hybridization Oven | VWR International | 230401V (Model 5420) | |
| Incubator Shaker | New Brunswick Scientific | Model Classic C25 | |
| KCl | Fisher Scientific | P217-500 | |
| Kimwipes | Kimerbly-Clark Professional | 34155 | Used for the humidity chamber |
| KH2PO4 | Fisher Scientific | P285-500 | |
| Microscope | Several commercial brands available | Any standard microscope capable of at least ~1.65X magnification is sufficient. | |
| Microscope slides | Fisher Scientific | 12-544-7 | |
| Mouse anti-hamsterSCP3 | Abcam | ab97672 | Does not work in our hands |
| Mouse anti-hMLH1 | BD Biosciences | 550838 | Does not work in our hands |
| Mouse anti-hRPA | Sigma-Alrich | MABE285 | Does not work in our hands |
| Na2HPO4 · 7 H2O | Fisher Scientific | S373-500 | |
| NaCl | Fisher Scientific | S271-3 | |
| Photo-Flo 200 solution | Electron Microscopy Sciences | 74257 | |
| Plastic transfer pipettes | Several commercial brands available | ||
| PNA TelC-Alexa647 | PNA Bio Inc | F1013 | Prepare as per manufacturer's instructions. |
| PNA TelC-Cy3 | PNA Bio Inc | F1002 | Prepare as per manufacturer's instructions. |
| ProLong Diamond Antifade Mountant | ThermoFisher Scientific | P36970 | |
| ProLong Diamond Antifade Mountant with DAPI | ThermoFisher Scientific | P36971 | |
| Rabbit anti-hRPA | Bethyl | A300-244A | Use at 1:300 |
| Rabbit anti-hSCP3 | Abcam | ab150292 | Use at 1:200 |
| Rabbit anti-hRad51 | GeneTex | GTX100469 | Use at 1:300 |
| Sodium citrate | Fisher Scientific | S279-500 | |
| Sucrose | Fisher Scientific | S5-500 | |
| Supercut Scissors, 30° angle, 10 cm | Fisher Scientific | 50-822-353 | Can also use any pair of small scissors. |
| Sylgard kit | Fisher Scientific | NC9897184 | Prepare as per manufacturer's instructions. |
| Triton X-100 | Fisher Scientific | BP151-100 | Dilute in sterile distilled water to make a 20% working solution. Store at room temperature. Triton X-100 forms a precipitate when diluted in water; precipitate dissolves overnight. |
| Trypsin | Worthington Biochemical | LS003708 | |
| Trypsin inhibitor from chicken egg white | Sigma-Aldrich | T9253-500MG | |
| Tween 20 | Bio-Rad | 170-6531 | Dilute in sterile distilled water to make a 20% working solution. Store at room temperature. |
| Vannas Spring Scissors – 4 mm (micro scissors) | Fine Science Tools | 15018-10 |
参考文献
- Nagaoka, S. I., Hassold, T. J., Hunt, P. A. Human aneuploidy: mechanisms and new insights into an age-old problem. Nature Reviews Genetics. 13, 493-504 (2012).
- Zickler, D., Kleckner, N. Recombination, Pairing, and Synapsis of Homologs during Meiosis. Cold Spring Harbor Perspectives in Biology. 7, (2015).
- Blokhina, Y. P., Nguyen, A. D., Draper, B. W., Burgess, S. M. The telomere bouquet is a hub where meiotic double-strand breaks, synapsis, and stable homolog juxtaposition are coordinated in the zebrafish, Danio rerio. PLoS Genetics. 15, e1007730 (2019).
- Saito, K., Sakai, C., Kawasaki, T., Sakai, N. Telomere distribution pattern and synapsis initiation during spermatogenesis in zebrafish. Developmental Dynamics. 243, 1448-1456 (2014).
- Oliver-Bonet, M., Turek, P. J., Sun, F., Ko, E., Martin, R. H. Temporal progression of recombination in human males. Molecular Human Reproduction. 11, 517-522 (2005).
- Gruhn, J. R., Rubio, C., Broman, K. W., Hunt, P. A., Hassold, T. Cytological studies of human meiosis: sex-specific differences in recombination originate at, or prior to, establishment of double-strand breaks. PLoS One. 8, e85075 (2013).
- Pratto, F., et al. Recombination initiation maps of individual human genomes. Science. 346, 1256442 (2014).
- Brown, P. W., et al. Meiotic synapsis proceeds from a limited number of subtelomeric sites in the human male. American Journal of Human Genetics. 77, 556-566 (2005).
- Poss, K. D., Nechiporuk, A., Stringer, K. F., Lee, C., Keating, M. T. Germ cell aneuploidy in zebrafish with mutations in the mitotic checkpoint gene mps1. Genes and Development. 18, 1527-1532 (2004).
- Saito, K., Siegfried, K. R., Nüsslein-Volhard, C., Sakai, N. Isolation and cytogenetic characterization of zebrafish meiotic prophase I mutants. Developmental Dynamics. 240, 1779-1792 (2011).
- Sansam, C. L., Pezza, R. J. Connecting by breaking and repairing: mechanisms of DNA strand exchange in meiotic recombination. Febs Journal. 282, 2444-2457 (2015).
- Feitsma, H., Leal, M. C., Moens, P. B., Cuppen, E., Schulz, R. W. Mlh1 Deficiency in Zebrafish Results in Male Sterility and Aneuploid as Well as Triploid Progeny in Females. 遺伝学. 175, 1561-1569 (2007).
- Dia, F., Strange, T., Liang, J., Hamilton, J., Berkowitz, K. M. Preparation of Meiotic Chromosome Spreads from Mouse Spermatocytes. Journal of Visualized Experiments. (129), e55378 (2017).
- Moens, P. B. Zebrafish: chiasmata and interference. Genome. 49, 205-208 (2006).
- Lisachov, A. P., Zadesenets, K. S., Rubtsov, N. B., Borodin, P. M. Sex chromosome synapsis and recombination in male guppies. Zebrafish. 12 (2), 174-180 (2015).
- Ocalewicz, K., Mota-Velasco, J. C., Campos-Ramos, R., Penman, D. J. FISH and DAPI staining of the synaptonemal complex of the Nile tilapia (Oreochromis niloticus) allow orientation of the unpaired region of bivalent 1 observed during early pachytene. Chromosome Research. 17 (6), 773 (2009).
