A representative result for amino acid-level signal to noise analysis for KCNQ1 is depicted in Figure 6. In this example, rare variants identified in the GnomAD cohort (control cohort), incidentally-identified WES variants (experimental cohort #1), and LQTS case-associated variants deemed likely disease-associated (experimental cohort #2) are depicted. Further, the signal-to-noise analysis comparing the WES and LQTS cohort variant frequency normalized against GnomAD variant frequency is depicted. LQTS-associated variants demonstrated high signal-to-noise ratios in domains corresponding with the channel pore, selectivity filter, and the KCNE1-binding domain. In comparison, incidentally identified variants in the WES cohort did not clearly demonstrate specific regions of high signal-to-noise elevation, suggesting that these variants reflect background genetic variation. This example did not utilize variant MAFs as noted above; however, it demonstrates all of the same principles as described.
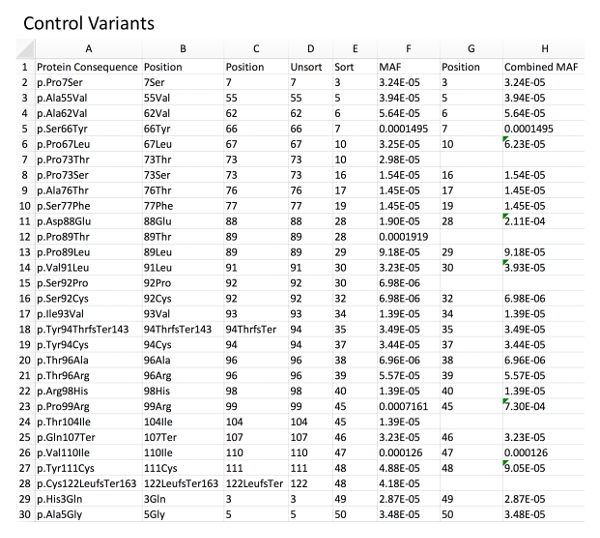
Figure 1: Example of control variant database with MAF calculation. Column A, directly imported GnomAD control rare variants. Column B, deletion of left-sided, non-position-related text from the variant nomenclature using an example formula for character removal (i.e.: for B2 "=RIGHT(A2,LEN(A2)-5", see Table of Materials). Column C, deletion of right-sided, non-position-related text from the variant nomenclature using a related formula (i.e.: for C2 "=LEFT(B2,LEN(B2)-3"). Column D, resultant unsorted amino acid positions. Column E, amino acid positions sorted in an ascending fashion to allow for identification of duplicate positions. Column F, associated MAF for each variant as imported from GnomAD. Column G and H, combined MAF for a given amino acid position (sum of each variant MAF at a specific position). Please click here to view a larger version of this figure.
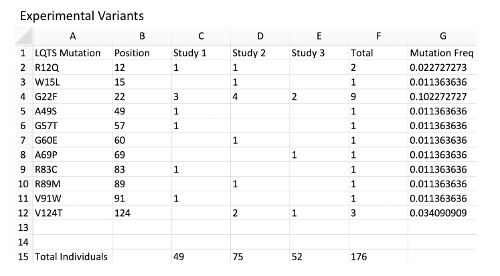
Figure 2: Example of experimental variant database with MAF calculation. Column A, a list of mock LQTS-associated mutations in KCNQ1 representing a disease-associated mutation experimental database. Column B, mutation position corresponding to each variant. Column C, a count of mutation-positive individuals within mock Study 1. Each are presumed to be heterozygous mutation carriers. The total number of individuals genotyped in the study is located at the bottom of the sheet. Column D, count of mutation-positive individual in mock Study 2. Column E, count of mutation-positive individual in mock Study 3. Column F, total mutation-positive individuals hosting the observed mutation across all studies. Note that distinct mutations associated with the same amino acid position should be combined. Column G, MAF of each mutation and amino acid position using an example formula (i.e.: for G2 "=2/(176*2) ", see Table of Materials). Note that since all individuals are presumed to be heterozygous and each individual presumed to carry 2 alleles of the KCNQ1 locus, the total individuals should be multiplied by 2 for the allele frequency. Please click here to view a larger version of this figure.
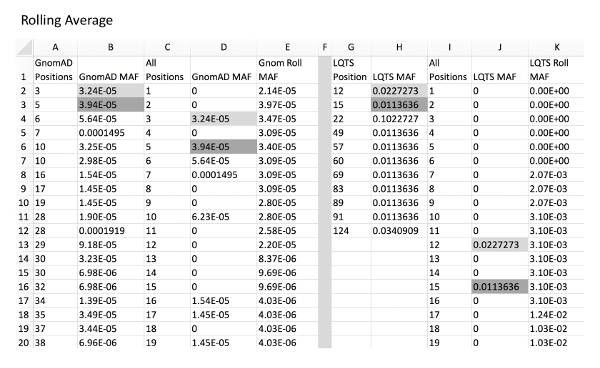
Figure 3: Example of rolling average calculation for control and experimental variants. Column A and B, GnomAD control variant positions and respective MAFs. Column C, all amino acid positions of KCNQ1 from amino acid position to final. Column D, GnomAD variant MAF for all positions with a MAF of 0 in place of positions without a variant. This can be automatically calculated using a VLOOKUP function (i.e. for D2, "=IFERROR(VLOOKUP(C2,A:B,2,),0), see Table of Materials). Column E, rolling average of position MAF using an example formula (i.e. for E2, "=SUM(D2:D7)/6" and for E7, "=SUM(D2:D12)/11"). Column G and H, LQTS experimental variant positions with respective MAFs. Column I, all amino acid positions of KCNQ1. Column J, LQTS variant MAF for all positions. Column K, rolling LQTS MAF. Gray fill cells are examples of where MAF values from columns B and H are expanded into column D and J, respectively, which correlate with respective positions in column C/I. Note that it is critical that all cells are formatted as "Numbers" for proper formula functioning. Please click here to view a larger version of this figure.
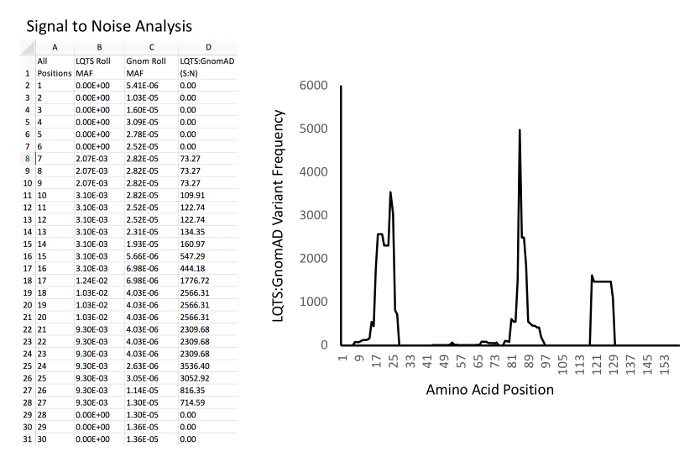
Figure 4: Example of signal-to-noise analysis and graphing. Left, example database and calculations. Column A, all amino acid positions of KCNQ1. Column B, LQTS experimental MAF rolling average for each position. Column C, GnomAD control MAF rolling average for each position. D: Signal-to-noise ratio (i.e. for D2, "=B2/C2"). Right, example of graph of signal-to-noise ratio (Y-axis) versus amino acid position (X-axis). Please click here to view a larger version of this figure.
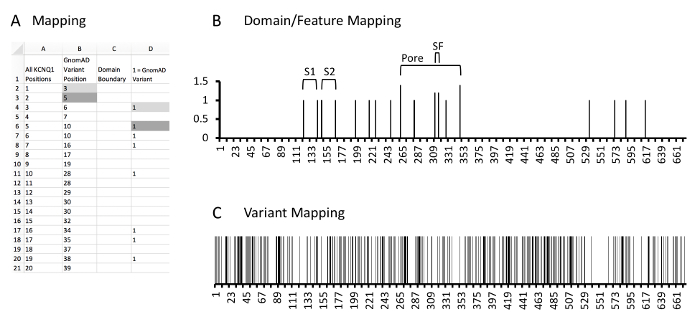
Figure 5: Example of protein and variant position mapping. A, example database and calculations. Column A, all amino acid positions of KCNQ1. Column B, KCNQ1 positions which have a rare control variant identified in GnomAD. Column C, the domain mapping column where cells containing values correspond to the N or C-terminal aspect of identified KCNQ1 protein domains or features. As the most N-terminal domain is the S1 domain has the N-terminal boundary at amino acid 122, no values are noted here. Column D, the variant mapping column where cells containing a 1 correspond to KCNQ1 positions which localize rare variants. Gray fill cells are two examples of where variant positions in column B are expanded into column D which correlate with respective positions in column A. Please click here to view a larger version of this figure.
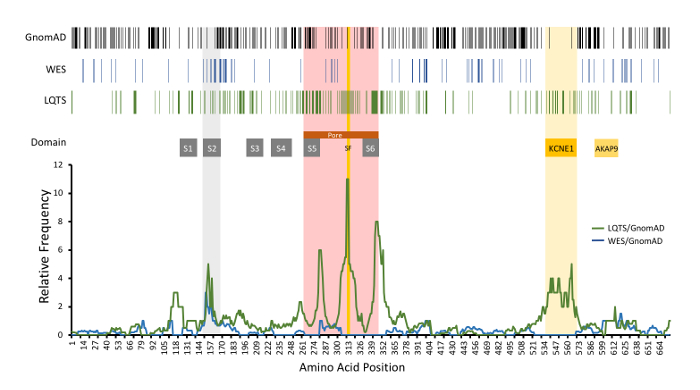
Figure 6: Example of amino acid-level signal-to-noise analysis of KCNQ1-encoded KCNQ1 (Kv7.1). Top, variant positions are demonstrated with vertical lines, including rare GnomAD cohort variants (black), incidentally-identified variants in WES referrals (blue), and variants identified in LQTS cases(green). Functional domains are noted. Relative frequency of LQTS case variants normalized to GnomAD variants (green line) is depicted compared to WES (blue line). S1-S6, transmembrane domains; SF, ion selectivity filter; KCNE1 and AKAP9, respective protein binding domains. Modified and reprinted with permission from previous work14. Please click here to view a larger version of this figure.
