Identification of major T cell subsets in spleen and thymus
In brief, single cell suspensions from spleen and thymus were lysed of red blood cells, incubated with 2.4G2 supernatant, followed by organelle-specific dye and surface marker staining with fluorescence-conjugated antibodies (Table 1). The developmental progression of thymocytes can be described by using antibodies to the co-receptors CD8 and CD4. The most immature thymocytes do not express CD4 or CD8 and are called double negative (DN). Then, they up-regulate both CD4 and CD8 and become double positive (DP). Finally, they down-regulate CD4 or CD8 and become mature single positive (SP) cells ready to leave the organ. The earliest stages of T cell development, when the cells do not express the co-receptors CD4 and CD8, can be further classified on the basis of CD44 and CD25 expression. The earliest progenitors are CD44+ CD25– (DN1), and then they start expressing CD25 and become CD44+CD25+ (DN2) cells. After that, the cells down-regulate CD44 and become CD44–CD25+ (DN3) cells and, finally, they down-regulate CD25 as well to become CD44–CD25– (DN4) cells that are on the way to expressing CD4 and CD8 and becoming double-positive (DP) cells.
In the spleen, the T cells can be divided into cytotoxic CD8+ and helper CD4+ T cells. Both of these populations can be further divided into naive, effector and memory subsets based on CD62L and CD44 staining. The detailed gating strategy is shown in Figure 1.
Quantification of mitochondria mass in different T cell subsets
Mitochondria-specific dyes were used to measure the amounts of mitochondria in T cell populations. We found that the mitochondrial contents were the lowest in DN1, peaked at DN2-DN3, and then slightly decreased in DN4 and DP thymocytes and were even lower in CD4 and CD8 SP thymocytes (Figure 2A). However, CD4 or CD8 SP thymocytes had higher mitochondria staining MFI than splenic CD4 or CD8 T cells (Figure 2B). These observations suggest that mitochondrial content fluctuates during T cell development with immature thymocytes containing more mitochondria than their mature counterparts do, and the naive T cells that recirculate in oxygen-rich blood have even lower mitochondrial mass. Interestingly, this reduction of mitochondrial contents was more prominent in the CD8 than the CD4 T lineage (Figure 2B), and T cell activation further decreased it. Among activated T cells, CD4+ memory T cells had slightly more mitochondria compared to effectors; however, the opposite was the case for CD8+ T cells: CD8+ memory T cells had the lowest mitochondrial mass among all T cell subsets (Figure 2B).
Lysosomal content measurement in various subpopulations of T cells
Using lysosome-specific dye, we observed relatively low, but detectable, lysosomal contents in all T cell populations, with more prominent presence of lysosomes in DN1 thymocytes (Figure 3A). No significant difference was found among the thymocyte subsets from DN1 onward. In the periphery, a relatively high number of lysosomes was found in memory and effector CD8+ T cells. This phenomenon is in line with previous studies showing that activated CD8+ T cells increased their expression of lysosomal-associated membrane protein 1 (LAMP-1)21,22, reflecting their possession of lysosomal-cytotoxic granules (Figure 3B)
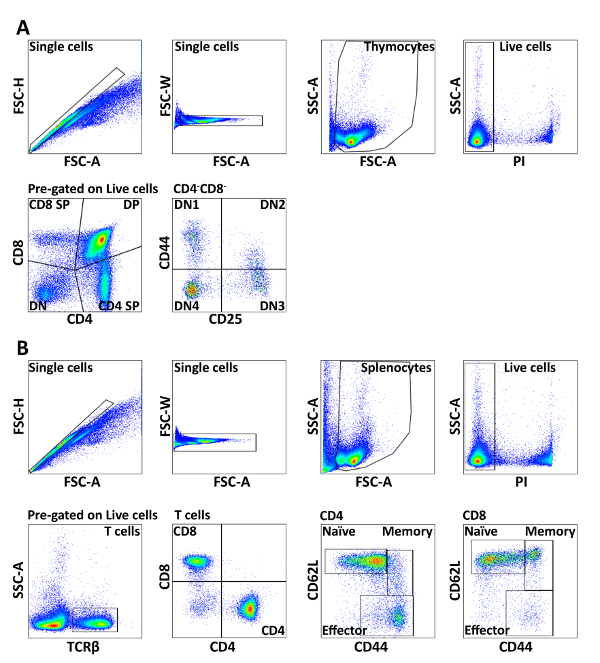
Figure 1: Gating strategy of splenic lymphocytes and thymocytes. Cells were pre-gated on FSC/SSC and PI– to obtain live singlets. (A) Total thymocytes from mice were stained with CD4, CD8, CD44, and CD25. CD4 and CD8 expressions were used to distinguish CD4+CD8– SP, CD4–CD8+ SP, CD4+CD8+ DP, and CD4–CD8– DN subsets. The CD4–CD8– DN subset was further divided into CD44+CD25– DN1, CD44+CD25+ DN2, CD44–CD25+ DN3, and CD44–CD25– DN4 subpopulations. (B) Total splenocytes from mice were stained with TCRβ, CD4, CD8, CD44, and CD62L. The TCRβ+ cells were separated into CD4+ and CD8+ T cells, which were further divided into naive (CD44–CD62L+), memory (CD44+CD62L+), and effector (CD44+CD62L–) populations. The results are representative of three independent experiments with n = 3-4. Please click here to view a larger version of this figure.
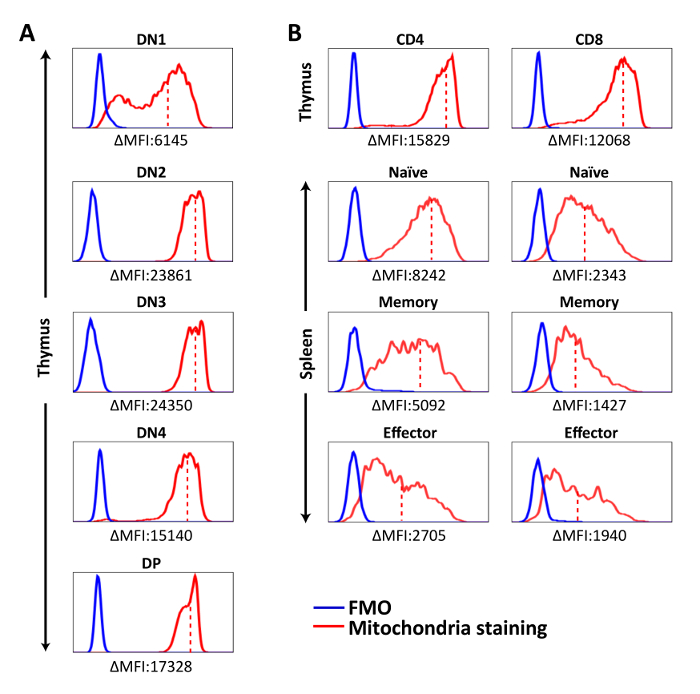
Figure 2: Representative histograms showing mitochondria staining in each cell population. Cells were stained with mitochondria-specific dye for 15 min at 37 ˚C, followed by surface marker staining. Fluorescent signal of stained cells was acquired by flow cytometer and analyzed on. (A) Mitochondria staining of DN and DP thymocytes. (B) Mitochondria staining of CD4SP and CD8SP thymocytes and splenic T cell populations. ΔMFI = MFI (Mitochondria staining) – MFI (FMO). The solid red line represents mitochondria staining, the solid blue line shows FMO control, and the dashed red line represents mean fluorescence intensity of mitochondria staining in each population. The results are representative of three independent experiments with n = 3-4. Please click here to view a larger version of this figure.
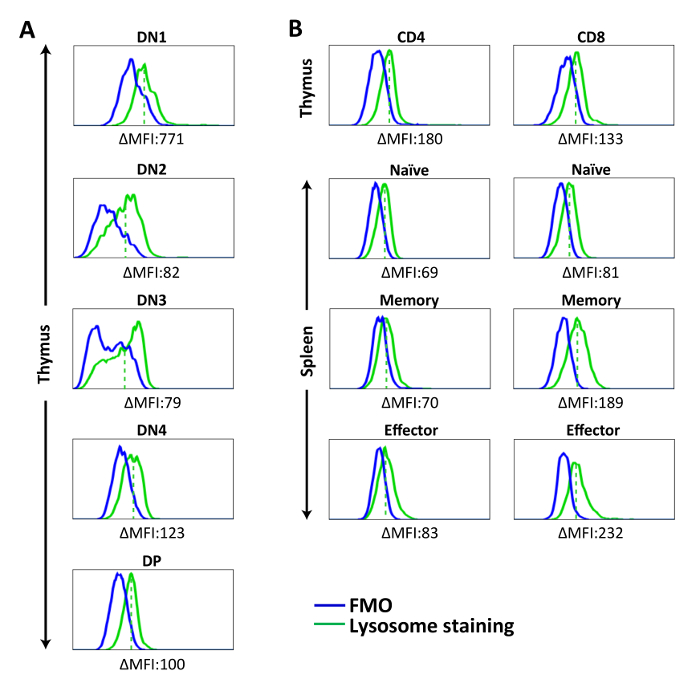
Figure 3: Representative histograms showing lysosome staining in splenic lymphocytes and thymocytes. Cells were stained with lysosome-specific dye for 30 min at 37 ˚C, followed by surface marker staining. Fluorescent signal of stained cells was acquired by flow cytometer and analyzed. (A) Lysosome staining of DN and DP thymocytes. (B) Lysosome staining of CD4SP and CD8SP thymocytes and splenic T cell populations. ΔMFI = MFI (Lysosome staining) – MFI (FMO). The solid green line represents lysosome staining, the solid blue line shows FMO, and the dashed red line represents mean fluorescence intensity of lysosome staining in each population. The results are representative of one experiment with n = 5. Please click here to view a larger version of this figure.
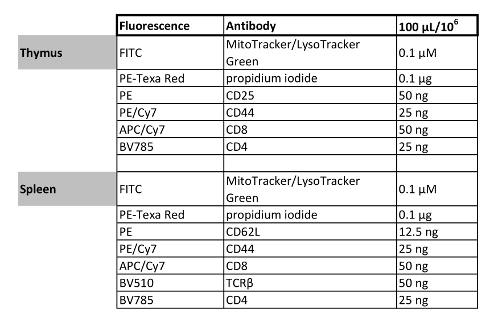
Table 1: Multicolor flow cytometry panel designs. The fluorochrome and concentration of antibodies/organelle dyes in the staining mixtures are listed here. It is strongly recommended to titrate and test each antibody when setting up new staining panels.
