Average brain volumes for 20 control participants, along with demographic information, is shown in Table 1. This acts as a guide for expected values when using these tools. Results should be viewed in the context of the original T1.nii image. All GM regions should be inspected as per the steps described in section 8. When performing visual QC, it is important to directly compare the GM regions to the T1 scan by viewing them overlaid on the T1.
Regions should be rejected for gross errors as shown in Figure 1. Sometimes these errors result if processing was run incorrectly, or if the brain was poorly positioned within the field of view. To correct these errors, the native T1 scans can be rigidly re-aligned to standard space and segmentation can be re-attempted. The rate of failures will vary depending on quality of the data and tools used, as well as the classification of failure. In the current study, failure rates of total failures resulting in rejection were < 5% for all tools, but less significant errors were consistently seen across a number of tools. FSL FAST, SPM 8 New Segment and FreeSurfer had errors (but not failures) in > 50% of scans for this cohort. This error rate was quantified by examining the notes taken during the visual QC process, with errors included if they were seen as a reasonable departure from the expected regions, as shown in Figures 2-6. It is important to note that these tools have been validated on other datasets and result in much lower error rates 3,8. While these errors could possibly be improved via manual intervention or inclusion of a mask at brain extraction, since SPM New Segment and MALP-EM resulted in a lower error rate for this dataset, these tools would be used instead. Masks can be applied before processing within ANTs and MALP-EM, and after processing for SPM (all versions) and FSL FIRST.
More minor errors are shown in Figures 2-6. By testing different segmentation tools on a dataset before application to the whole cohort, the tool that performs best on that dataset can be selected for analysis. When performing QC, a procedure should be developed for choosing to reject, edit, or accept segmentations. Common errors seen for the seven tools are described here, with examples shown in Figures 2-6. Errors in segmentation such as these can often be corrected with the addition of a mask in the processing stream or editing the regions. However, regions with extensive over- or under-estimation of the cortex may need to be rejected from analysis. Strict criteria should be developed and followed when making this decision. These steps are not covered in this protocol and will vary from dataset to dataset.
Generally, when performing visual QC, it is important to pay particular attention to temporal and occipital regions, as these are areas that show the most consistent errors. Figure 2 shows examples of good and bad temporal segmentations, and Figure 3 shows examples of good and bad occipital segmentations. Figure 4 shows another common issue that occurs in all tools, in which non-brain tissue is classified as CGM in superior slices of the brain. Figure 5 displays another issue seen in a number of segmentations where regions of the CGM are excluded from the segmentation. This often occurs in superior slices of the brain, as seen in Figure 5.
SPM8 Unified Segment commonly resulted in poor temporal delineation, with the segmented GM region spilling into non-brain tissue surrounding the temporal lobes. Spillage into the occipital lobe is common, while under-estimation of the frontal lobes also seen in a number of regions. For SPM8 New segment, poor temporal delineation and occipital spillage were also common. Using this version of SPM also results in voxels within the skull and dura being classified as GM in nearly all segmentations. SPM12 was improved compared to earlier versions of SPM, with the temporal lobe segmentations improved and less spillage in other regions. ANTs showed highly variable performance on this cohort, with the initial brain extraction determining the quality of segmentation. It is important to pay particular attention to the external boundaries, and if brain extraction is poor using ANTs, then the brain mask included in the Atropos command can be improved. Issues with over-estimation of the GM in the temporal and occipital lobes were again common. MALP-EM showed fewer issues with over-estimation of the temporal and occipital lobes; although, there was under-estimation of the cortex in a number of cases. This can be improved by inclusion of a brain mask in the pipeline. FSL FAST segmentations were highly variable, due to the variable performance of BET brain extraction on the data from this cohort. Again, issues within occipital and temporal lobes were common; however, these can be improved with optimization of brain extraction. Finally, FreeSurfer volumetric regions are often tight along the GM/CSF boundary, typically excluding some regions of GM in the outer boundary (Figure 6). As with other tools, spillage outside of the GM is prevalent within the temporal and occipital lobes. Finally, Figure 7 shows an example of a good segmentation displayed in FSLview that had no errors in segmentation. Manual editing of the regions can often be performed to improve regions, although this is not covered here.
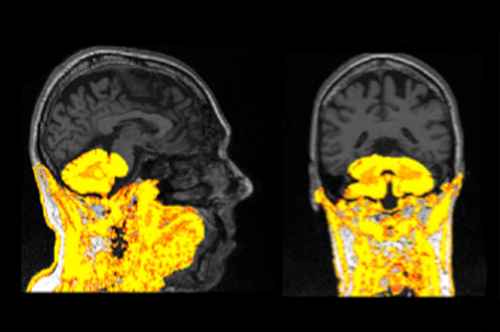
Figure 1: Example of a failed segmentation displayed on a T1 scan. This segmentation should be re-processed and excluded from analysis if it cannot be improved. Please click here to view a larger version of this figure.
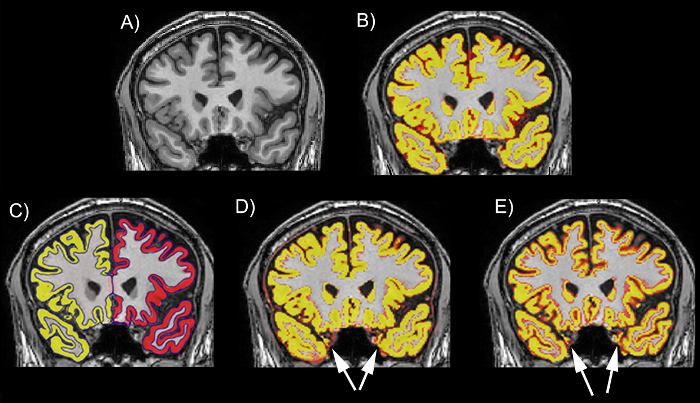
Figure 2: Examples of the performance of different tools on the temporal lobe on a T1 scan. (A) The T1 scan without a segmentation. (B) The T1 scan with an example of a good regional delineation (MALP-EM). (C) The T1 scan with an example of a good regional delineation (FreeSurfer). (D) The T1 scan with an example of a poor regional delineation, showing spillage in the left and right temporal lobes (SPM 8 New Segment). (E) The T1 scan with an example of a poor regional delineation, showing spillage in the left and right temporal lobes (FSL FAST). The scans are viewed in FSLeyes with the T1 scan as a base image and the GM region as an overlay. In this figure, the GM regions are viewed as red-yellow with an opacity of 0.4. The color gradient represents partial volume of voxels, with voxels that are more yellow having a higher PVE estimate (more likely to be GM) and those that are red having a lower PVE estimate (less likely to be GM). Please click here to view a larger version of this figure.
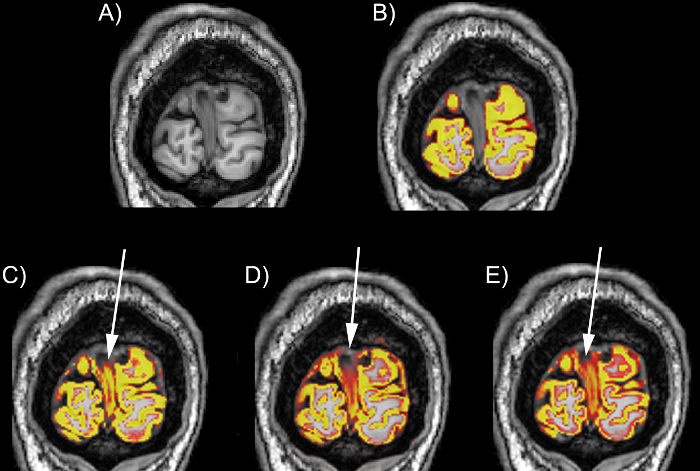
Figure 3: Examples of the performance of different tools on the occipital lobe on a T1 scan. (A) The T1 scan without a segmentation. (B) The T1 scan with an example of a good regional delineation (MALP-EM). (C) The T1 scan with an example of a poor occipital lobe delineation with spillage into the dura in the medial section of the region (SPM 8 Unified Segment). (D) The T1 scan with an example of a poor occipital lobe delineation with spillage into the dura in the medial and superior sections of the region (SPM 8 New Segment). (E) The T1 scan with an example of a poor occipital lobe delineation with spillage into the dura in the medial and superior sections of the region (FSL FAST). The scans are viewed in FSLeyes with the T1 scan as a base image, and the GM region as an overlay. In this figure, the GM regions are viewed as red-yellow with an opacity of 0.4. The color gradient represents partial volume of voxels, with voxels that are more yellow having a higher PVE estimate (more likely to be GM) and those that are red having a lower PVE estimate (less likely to be GM). Please click here to view a larger version of this figure.

Figure 4: Example of a GM region spilled into the dura, displayed in an FSLview window (in sagittal, coronal, and axial views). The blue region highlights spillage into the dura. Please click here to view a larger version of this figure.
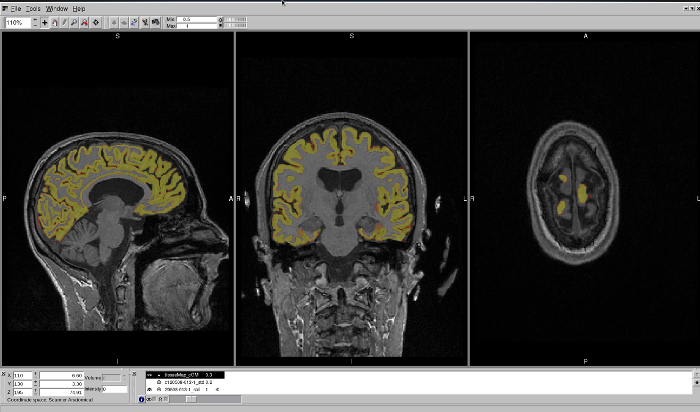
Figure 5: Example of a GM region that has excluded regions of the CGM from segmentation. This region is displayed in an FSLview window, in sagittal, coronal, and axial views. The axial view best shows the regions that have been excluded from segmentation. Please click here to view a larger version of this figure.
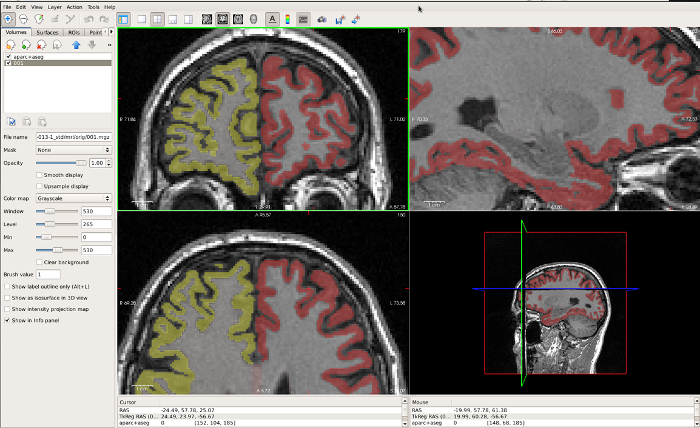
Figure 6: Example of a FreeSurfer GM region that is very tight along the GM/CSF boundary, displayed in FreeView. The coronal window in the top left best displays the under-estimation in the CGM in this region. Please click here to view a larger version of this figure.
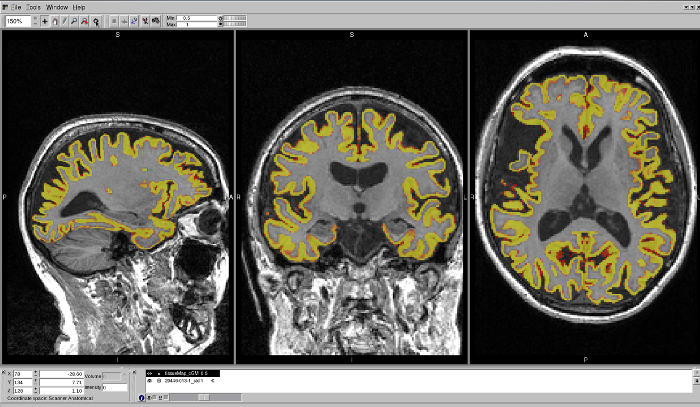
Figure 7: Example of a well-delineated MALP-EM region on a T1 brain scan. The region shows no issues with over- or under-estimation of the CGM in any region. Please click here to view a larger version of this figure.
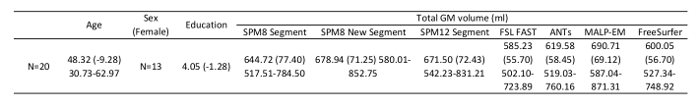
Table 1: Demographic information and average GM volumes (mL) for 20 control participants from the TRACK-HD study, segmented using the seven tools described here.
