The efficiency of the protocoll was demonstrated by genotyping the offspring of an heterozygous cross (aldh7a1+/ot100, here shown as aldh7a1+/-) (Figure 3A-C)9. The aldh7a1ot100 mutant allele has a 5-base pair (bp) insertion in the first coding exon of the zebrafish aldh7a1 gene that leads to frameshift and loss-of-function due to an early stop codon9. Four primers were used in a multiplex reaction to obtain three distinctive bands to differentiate between WT, heterozygous (aldh7a1+/-), and homozygous mutant (aldh7a1-/-) genotypes (from Pena, et al.9): Gen1_FW 5'-ATGATGCAGCGCGTGCTGAC-3', "Gen2_RV":5' -CCCTTTGAACCTCACAGGAGTT-3', "5 nt-ins-specific_FW":5'-TGTTTTCAACGGTTCAACGG-3', and "WT-specific_RV":5'-TCCCTGTCCTCCCCAAGAAC-3'). The amplification of both alleles using the Gen1_FW and Gen1_RV primers resulted in a 434-bp amplicon. A 293-bp band arose from specific amplification of the mutant allele, and a 195-bp band was obtained specifically for the WT allele, as shown in Figure 3A. Following PCR, 8 µL of each 20 µL reaction volume was analyzed on a 1% agarose/sodium borate gel, as shown in Figure 3B. Sufficient larval DNA was recovered from 98% (n = 576) of the samples, resulting in a high PCR efficiency rate. DNA extraction using the NaOH technique obtains on average 4.7 0.5 ng/µL of larval DNA per sample following the fin biopsy procedure, in ~30 µL volume. DNA extraction using chelating resin results in a similar yield, on average 3.8 0.5 ng/µL of DNA per sample. This amount of DNA collected from larval fin transections generates PCR-ready genomic DNA of sufficient quality to allow for the identification of genotypes. Each DNA sample can be used in ˜30 PCR amplification reactions. Amplification products obtained using primers Gen1_FW and Gen2_RV can also be used for sequencing applications9, which successfully identified the 5-bp insertion in the homozygous mutants (aldh7a1-/-) (Figure 3C). Sanger sequencing was performed via an external service to test if the PCR products were suitable for this application. DNA samples from agarose-gel confirmed homozygous mutants and WTs (n = 3 each) were used. High Phred19 scores (>30) were obtained indicating high quality reads, except for the first and last 40 base pairs.
This protocol was subsequently used to genotype various other zebrafish mutations by heteroduplex melting analysis. The plpbpot101 and plpbpot102 mutant alleles of the plpbp gene (Figure 4A-D) each lead to a frameshift and early stop codon. In order to identify plpbp-null zebrafish larvae, two primers were used to amplify the first coding exon including the mutation site (plpbp-F 5'-GCACTCTGGCTATGTGGAGA-3' and plpbp-R 5'-AGCTGTCACTCATCCCTCGT-3'). The amplification of the plpbp fragment in WT animals results in a 272-bp amplicon (homoduplex). The amplicon obtained from plpbpot101 (4-bp deletion allele, Figure 4C) consists of a fragment of 268-bp and that of the plpbpot102 (5bp-substitution, 2-bp deletion allele, Figure 4D), 270-bp, as seen in Figure 4A-B. After denaturation and annealing, the PCR fragments from heterozygous animals would contain heteroduplex and homoduplex DNA17. Homoduplex and heteroduplex bands can be easily separated and visualized using polyacrylamide gel electrophoresis (PAGE). The presence of an open angle between matched and mismatched DNA strands caused by imperfect annealing due to the occurrence of mutations cause the heteroduplex DNA to migrate at a significantly slower pace than homoduplex DNA17. Distinct migration patterns are produced by distinct mutations. The heterozygous genotypes (plpbp+/ot101, plpbp+/ot102) would therefore display heteroduplex DNA fragments, as well as the compound heterozygous (plpbpot101/ot102) (Figure 4A-B). Precise genotyping is obtained by this heteroduplex melting assay as a unique PAGE pattern is obtained for each genotype and distinct heterozygous mutations can be visualized (Figure 4A-B). Homozygous mutants would display a homoduplex pattern in the PAGE gel and therefore be indistinguishable to WTs. To distinguish these genotypes, another round of PAGE gel analyses should be performed in which PCR products from WT alleles need to be mixed with the samples to be studied, as described elsewhere17.
About 90% (88/98) of WT and heterozygous embryos were successfully raised to adulthood (>2 months). As aldh7a1 and plpbp are loss-of-function mutants that display an early death phenotype, untreated animals could not be raised to adulthood. However, the genotypes of all surviving WT and heterozygous adults were identical to larval fin clipping results, indicating a 100% accuracy rate for the identification of genotypes using the larval fin biopsy procedure.
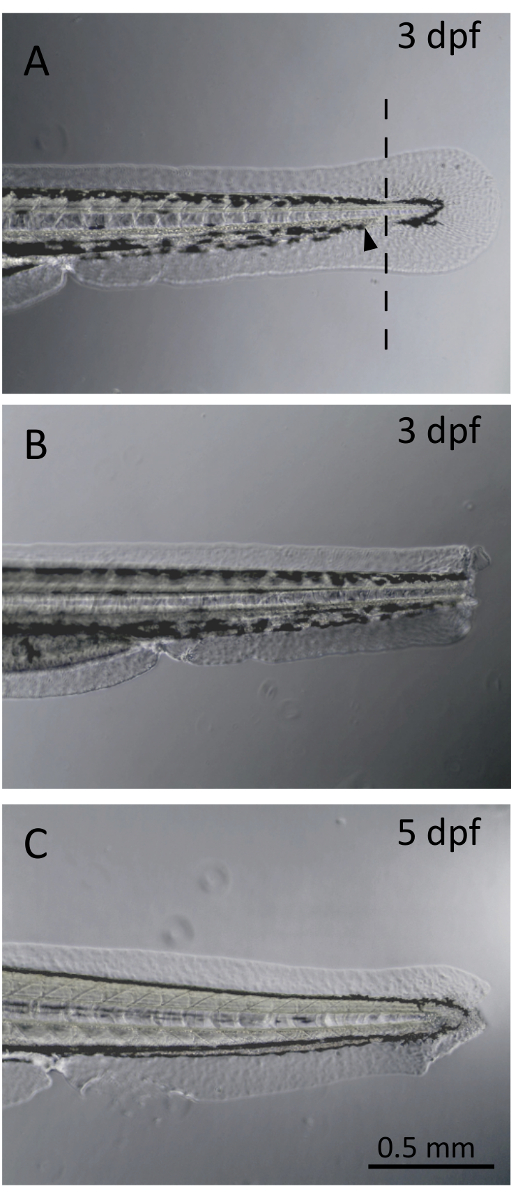
Figure 1. Larval fin clip. (A). Non-severed larval fin at three days post-fertilization (dpf). The line represents the site of transection, located within the pigment gap. The black arrowhead demonstrates the limit of the caudal blood circulation. (B). Larva following fin clipping procedure at 3 dpf. (C). Larval zebrafish fin regrowth at 5 dpf. Fin regeneration from a blastemal formation is observed only 2 days post-fin transection. Please click here to view a larger version of this figure.

Figure 2. Procedure for larval fin clipping. (A). The larvae are placed onto the tape and excess embryo media is removed. Under the microscope, the fin is transected at the region of the pigment gap as shown in Figure 1A. Using a microscalpel, the fin is collected and placed onto a filter paper surface simply by touch (B), and can be readily visualized as a black spot. Hold the piece of filter paper containing the fin and cut a small square surrounding the desired region (C). Place the small piece of filter paper containing the fin into a well of a 96 well plate (D). The corresponding larva should be placed into the corresponding well of a flat-bottom 96-well plate in 200 µL of embryo media (E). The filter paper piece should be submerged in the lysis solution (F). Please click here to view a larger version of this figure.
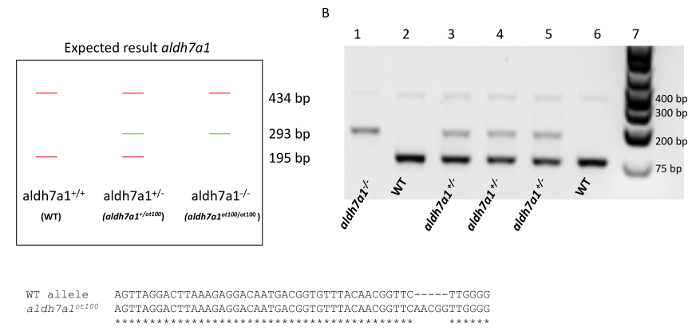
Figure 3. Genotyping of aldh7a1 heterozygous cross from larval fin biopsies. (A). Expected result where amplification of both WT and aldh7a1ot100 alleles results in a 434-bp amplicon. A 293-bp band arises from amplification of the mutant allele (aldh7a1ot100, 5-bp insertion), and a 195-bp band is obtained for the WT allele. (B). Example of PCR amplification from aldh7a1 larval fin tissues. A 1 kb molecular weight marker is shown in lane 7. Lanes 1 – 6 each represent a single fin biopsy. PCR results identify aldh7a1-/- (aldh7a1ot100/ot100) genotypes (lane 1), aldh7a1+/- heterozygous genotypes (aldh7a1+/ot100) (lanes 3, 4, and 5) and WT (aldh7a1+/+) genotypes (lanes 2 and 6). (C). Alignment between the sequenced WT and aldh7a1ot100 allele showing the position of the 5-bp insertion mutation. This figure was adapted from the Supplemental Figure 5 of Pena et al.9 with permission granted from the Genetics Society of America. Please click here to view a larger version of this figure.
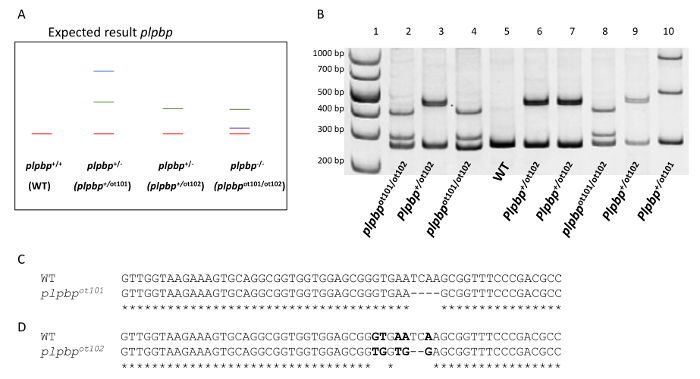
Figure 4. Genotyping of plpbp compound heterozygous (2 bp deletion /4 bp deletion) from larval fin biopsies. (A). Expected result where amplification of the WT allele resulted in a 272-bp amplicon. The 4-bp deletion allele (plpbpot101) and the 5-bp substitution with 2-bp deletion allele (plpbpot102) produce amplicon lengths of 268-bp and 270-bp respectively. A heteroduplex is formed between a WT allele and the mutant allele in a heterozygous genotype (plpbp+/-). Formation of a heteroduplex due to mismatching alleles will cause retardation of the band within the gel compared to the WT homoduplex. (B). PCR amplification resulting from larval fin clippings of the offspring of a plpbp+/ot101x plpbp+/ot102 cross. A 1 kb molecular weight marker is shown in lane 1. Lanes 2 – 10 each represent a single fin biopsy. PCR results identify mutant genotypes (plpbp-null compound heterozygous plpbpot101/ot102, lanes 2, 4, and 8), heterozygous genotypes (lanes 3, 6, 7, 9, and 10) and WT genotypes (lane 5). (C). Alignment between the sequenced WT and plpbpot101 allele showing the position of the 4-bp deletion mutation. (D). Alignment between the sequenced WT and plpbpot102 allele showing the position of the 2-bp deletion and 5-bp substitution (bold). Please click here to view a larger version of this figure.
| Components | 20 μl reaction | 100 reactions | Final Conc. |
| Nuclease-free H2O | 7.45 μL | 745 μL | |
| Go-Taq 2x (Promega, M7122) | 10 μL | 1000 μL | 1x |
| primer Gen1_FW | 0.25 μL | 25 μL | 0.125 μM |
| primer 5 nt-ins-specific_FW | 0.3 μL | 30 μL | 0.15 μM |
| Primer Gen2_RV | 0.25 μL | 25 μL | 0.125 μM |
| Primer WT-specific_RV | 0.25 μL | 25 μL | 0.125 μM |
| DNA | 1.5 μL | — |
Table 1. Reaction conditions for multiplex PCR used for the aldh7a1 allele in this protocol. The primers used are as follows: Gen1_FW 5'-ATGATGCAGCGCGTGCTGAC-3', "Gen2_RV":5' -CCCTTTGAACCTCACAGGAGTT-3', "5 nt-ins-specific_FW":5'-TGTTTTCAACGGTTCAACGG-3', and "WT-specific_RV":5'-TCCCTGTCCTCCCCAAGAAC-3'. Primer stocks were 10 µM.
| Components | 13 μl reaction | 100 reactions | Final Conc. |
| H2O | 3.25 μL | 325 μL | |
| Go-Taq 2x (Promega, M7122) | 6.25 μL | 625 μL | 1x |
| primer FW | 1 μL | 100 μL | 0.77 μM |
| Primer RV | 1 μL | 100 μL | 0.77 μM |
| DNA | 1.5 μL | — |
Table 2. Reaction conditions for PCR used for plpbp allele in this protocol. The forward primer used was PLPBP-F 5'-GCACTCTGGCTATGTGGAGA-3' and the reverse, PLPBP-R 5'-AGCTGTCACTCATCCCTCGT-3'. Primer stocks were 10 µM.
| Cycle Step | Temp. | Time | Cycles |
| Initial denaturation | 95 °C | 3 min | 1 |
| Denaturing | 95 °C | 30 sec | 36 |
| Annealing* | X °C | 30 sec | |
| Extension | 72 °C | 20 sec/kb | |
| Final Extension | 72 °C | 5 min | 1 |
| 4 °C | hold |
Table 3. PCR conditions used in this protocol.
