1. Manufacturing Thin Two-layered Polyacrylamide (PA) Hydrogels on Multi-Well Plates
- Dissolve ammonium persulfate (APS) in distilled ultrapure water to achieve a final concentration of 10 g/mL. Aliquot and store the solution at 4 °C for short-term use (3 weeks).
NOTE: The above solution can be prepared in advance of hydrogel fabrication. - Glass activation of 24-well dishes
- Incubate 24-well glass bottom plates with 13 mm-diameter wells (see Table of Materials) for 1 h with 500 µL of 2 M NaOH per well at room temperature.
- Rinse the wells 1x with ultrapure water and then add 500 µL of 2% (3-Aminopropyl)triethoxysilane (see Table of Materials) in 95% ethanol to each well for 5 min.
- Rinse the wells 1x again with water and add 500 µL of 0.5% glutaraldehyde to each well for 30 min. Rinse the wells 1x with water and dry them at 60 ˚C with the lid off.
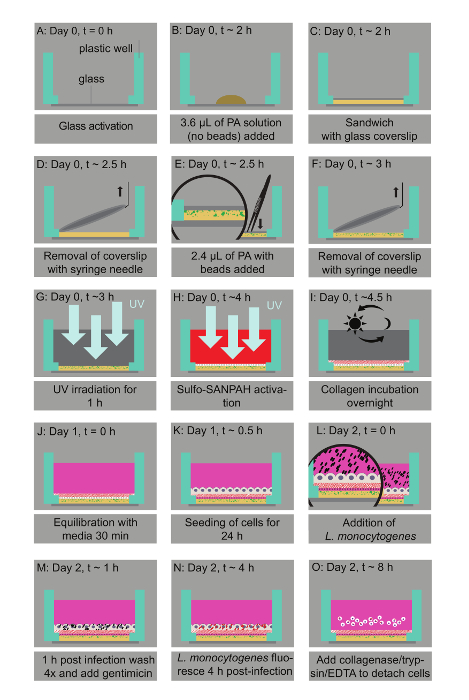
Figure 1: Bacterial infection assay of host cells residing on thin two-layered fluorescent bead-embedded polyacrylamide (PA) hydrogels of varying stiffness. A. Glass coverslips are chemically modified to enable hydrogel attachment. B. 3.6 µL of PA mixtures are deposited on the glass bottoms. C. The mixture is covered with a 12-mm circular glass coverslip to enable polymerization. D. The coverslip is removed with a needle syringe. E. 2.4 µL of a PA solution with microbeads is added on top of the bottom layer and capped with a circular glass coverslip. F. A buffer is added in the well and the coverslip is removed. G. UV irradiation for 1 h ensures sterilization. H. A Sulfo-SANPAH-containing solution is added on the gels, which are then placed under UV for 10 min. I. The hydrogels are washed with a buffer and then incubated overnight with collagen I. J. The hydrogel is equilibrated with cell media. K. The host cells are seeded. L. Lm bacteria are added to the solution and the infection is synchronized via centrifugation. M. 1 h post-infection bacteria in the solution are washed away and media supplemented with an antibiotic is added. N. At 4 h post-infection, Lm (JAT985) starts fluorescing. O. HMEC-1 cells are detached from their matrix and the solutions are transferred to tubes to perform flow cytometry measurements. Note that days and approximate times for each step of the assay are also indicated. This figure has been modified from Bastounis and Theriot59. Please click here to view a larger version of this figure.
- Polyacrylamide hydrogel fabrication
NOTE: See Figure 1.- Prepare aqueous solutions that contain 3 – 10% of 40% stock acrylamide solution (see Table of Materials) and 0.06 – 0.6% of 2% stock bis-acrylamide solution (see Table of Materials) to manufacture hydrogels of tunable stiffness ranging from 0.6 kPa to 70 kPa. See Table 1.
- For 0.6 kPa hydrogels, mix 3% acrylamide with 0.045% bis-acrylamide. For 3 kPa hydrogels, mix 5% acrylamide with 0.074% bis-acrylamide. For 10 kPa hydrogels, mix 10% acrylamide with 0.075% bis-acrylamide. For 20 kPa hydrogels, mix 8% acrylamide with 0.195% bis-acrylamide. For 70 kPa hydrogels, mix 10% acrylamide with 0.45% bis-acrylamide.
NOTE: Further details on achieving the desirable PA hydrogel stiffness can be found elsewhere24,25,26,27.
- For 0.6 kPa hydrogels, mix 3% acrylamide with 0.045% bis-acrylamide. For 3 kPa hydrogels, mix 5% acrylamide with 0.074% bis-acrylamide. For 10 kPa hydrogels, mix 10% acrylamide with 0.075% bis-acrylamide. For 20 kPa hydrogels, mix 8% acrylamide with 0.195% bis-acrylamide. For 70 kPa hydrogels, mix 10% acrylamide with 0.45% bis-acrylamide.
- Prepare two aqueous solutions for each desirable hydrogel stiffness. Prepare Solution 1 to be bead-free and Solution 2 to contain 0.03% 0.1-µm diameter fluorescent micro-beads (see Table of Materials).
- Degas Solutions 1 and 2 by vacuum for 15 min to eliminate oxygen that is known to inhibit polymerization of the solutions.
- Add 0.6% of the 10 g/mL stock APS solution and 0.43% tetramethylethylenediamine (TEMED) to Solution 1 to enable a polymerization initiation. Act fast.
- Add 3.6 µL of Solution 1 to the center of each well of the 24-well dish (see step 1.2 for its preparation).
- Immediately cover the wells with 12-mm untreated circular coverslips and let Solution 1 sit for 20 min so that it fully polymerizes.
- Gently tap a syringe needle to a surface to create a small hook at its tip to facilitate the removal of the coverslips. Lift the coverslips using the syringe needle.
- Add 0.6% of the 10 g/mL stock APS solution and 0.43% TEMED to Solution 2. Deposit 2.4 µL of the mixture on top of the first layer in each well of the 24-well dish.
- Cover Solution 2 with 12-mm circular glass coverslips, gently pressing downwards using a pair of forceps to ensure the thickness of the second layer is minimal. Let Solution 2 polymerize for 20 min.
- Add 500 µL of 50 mM HEPES pH 7.5 to each of the wells and then remove the glass coverslips with the syringe needle and forceps.
- Prepare aqueous solutions that contain 3 – 10% of 40% stock acrylamide solution (see Table of Materials) and 0.06 – 0.6% of 2% stock bis-acrylamide solution (see Table of Materials) to manufacture hydrogels of tunable stiffness ranging from 0.6 kPa to 70 kPa. See Table 1.
- Sterilization, collagen-coating, and equilibration of polyacrylamide hydrogels
- UV-expose the hydrogels for 1 h in the tissue culture hood to allow sterilization.
- Prepare a mixture of 0.5% weight/volume sulfosuccinimidyl 6-(4′-azido-2′-nitrophenylamino)hexanoate (Sulfo-SANPAH, see Table of Materials) in 1% DMSO and 50 mM HEPES pH = 7.5.
- Add 200 µL of this solution to the upper surface of the hydrogels. Acting fast, expose them to UV (302 nm) for 10 min to activate them.
- Wash the hydrogels twice with 1 mL of 50 mM HEPES pH = 7.5. Repeat if needed to ensure that any excess crosslinker is removed.
- Protein-coat the hydrogels with 200 µL of 0.25 mg/mL rat tail Collagen I (see Table of Materials) in 50 mM of HEPES. Incubate the hydrogels, with the collagen solution on top, overnight at room temperature.
NOTE: To prevent dehydration/evaporation, place the multi-well plates in a secondary containment and add laboratory cleaning tissues soaked in water in the inner periphery of the containment. - Use an epifluorescence or confocal microscope with a 40X objective to measure the thickness of the hydrogels. Do so by locating the z positions of the bottom (where the glass surface is) and top planes of the hydrogel (where the fluorescent beads' intensity is maximum). Then subtract the z positions to determine the height.
NOTE: We used an inverted epifluorescence microscope and a 40X objective with numerical aperture 0.65 to measure the thickness of the hydrogels. Atomic Force Microscopy measurements (AFM) can also be performed at this point to confirm the exact stiffness of the hydrogels (see Figure 2). - Before seeding the cells of interest on the hydrogels, add 1 mL of media on the hydrogels and incubate them at 37 ˚C for 30 min to 1 h to ensure equilibration.
NOTE: We added MCDB-131 full media because that is the media where the model host cells (HMEC-1) were cultured in (see step 2.1 for details).
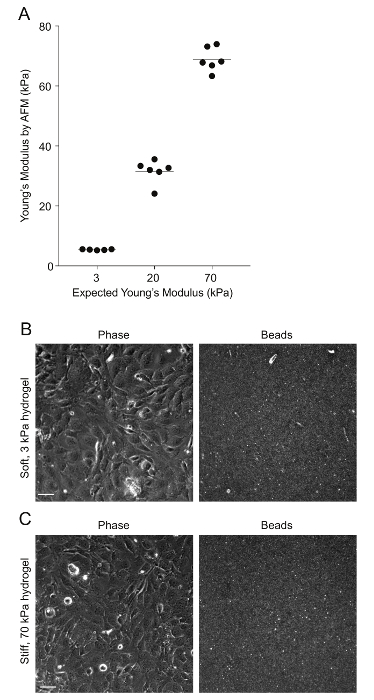
Figure 2: AFM measurements of PA hydrogel stiffness and beads' distribution. A. Data show the expected Young's modulus (measure of stiffness) of the PA hydrogels, given the amount of acrylamide and bis-acrylamide used versus the Young's modulus measured through AFM (N = 5 – 6). The horizontal bars depict the mean. The stiffness of the 0.6 kPa hydrogels could not be measured because the hydrogels were very soft and adhered to the AFM tip. B. This is a phase image of confluent HMEC-1 cells and the corresponding image of the beads embedded on the uppermost surface of a soft 3 kPa-PA hydrogel. The HMEC-1 were seeded for 24 h at a concentration of 4 x 105 cells per well. C. This image is the same as Figure 2B but for cells residing on a stiff 70-kPa PA hydrogel. Please click here to view a larger version of this figure.
2. Human Microvascular Endothelial Cell Culture and Seeding on Hydrogels
- Culture HMEC-1 (human, microvascular endothelial) cells in MCDB-131 full media containing MCDB-131 media supplemented with 10% fetal bovine serum, 10 ng/mL of epidermal growth factor, 1 µg/mL of hydrocortisone, and 2 mM of L-glutamine (see Table of Materials).
- Split the confluent cultures 1:6 every 3 – 4 days and keep the cells until passage 40.
- One day prior to the experiment, detach the cells from their culture vessel using 0.25% trypsin/EDTA. First wash the cells and their culture vessel 1x with sterile phosphate buffered saline (PBS), and then add the appropriate amount of 0.25% trypsin/EDTA (2 mL per 100-mm dish or 75-cm flask, 1 mL per 60-mm dish or 25-cm flask), incubating the flask at 37 °C for 5 – 10 min to allow the detachment of the cells from their substrate.
- Neutralize the trypsin by adding the desired volume of MCDB-131 full media, pipet gently to break up the clumps of cells, and then place the solution into a conical centrifuge tube.
- Gently swirl the solution of cells to ensure that the cells are evenly distributed and then take out 20 µL of the solution and very gently fill out the two chambers underneath the coverslip of a glass hemocytometer.
- Pellet down the solution of cells contained in the conical centrifuge tube using centrifugation for 10 min at 500 x g.
- During the 10 min waiting period, count the cells using a hemocytometer. Use a microscope, focus on the grid lines of the hemocytometer with a 10X objective, and then use a hand tally counter to count the number of cells in one 1 mm x 1 mm square.
- Move the hemocytometer to another 1 mm x 1 mm square, count the cells there and then repeat the process two more times. Calculate the average of the four measurements and then multiply the average by 104. The final value is the number of viable cells/mL in the cell suspension that is being centrifuged.
- Remove the liquid out of the conical centrifuge tube while ensuring that the cell pellet is not disrupted. Resuspend the cells in the MCDB-131 full media at a concentration of 4 x 105 cells/mL.
- Seed the cells in suspension on the hydrogels by first removing the media with which the hydrogels were incubated and then adding 1 mL of cell suspension on each hydrogel.
3. Infection of Human Microvascular Endothelial Cells with L. monocytogenes
- Advance preparation
Note: Prepare the following solutions in advance.- Streptomycin, chloramphenicol, and gentamicin stocks
- Prepare 50 mg/mL of streptomycin stock solutions by weighing 0.5 g of streptomycin sulfate and dissolving it completely into 10 mL of ultrapure water.
- Prepare 7.5 mg/mL of chloramphenicol stock solutions by weighing 75 mg of chloramphenicol and dissolving it completely into 10 mL of 100% ethanol.
- Prepare 20 mg/mL of gentamicin stock solutions by weighing 0.2 g of gentamicin sulfate and dissolving it completely into 10 mL of ultrapure water.
- Filter-sterilize all stock solutions with a 0.2-µm syringe filter and store them at -20 °C for long-term use (1 – 2 months).
- Brain heart infusion (BHI) media and agar plates
- Locate two 1-L flasks and add a magnetic stir bar inside each flask.
- For the BHI media, add 37 g of BHI powder in one flask and add ultrapure water up to 1 L. Mix the solution vigorously by placing the flask on a magnetic stir plate until the powder is dissolved.
- For the BHI agar plates, add 37 g of BHI powder and 15 g of granulated agar (see Table of Materials) in the second flask and add ultrapure water up to 1 L. Mix the solution vigorously by placing the flask on a magnetic stir plate until the powder is dissolved.
- Screw the lids of the flasks not too tightly and autoclave the solutions using the liquid setting or according to the autoclave's specifications.
- Remove the solution from the autoclave, then cool down the agar-BHI solution to 55 °C. If the BHI media in the 1 L flask is kept sterile, it can be used for up to a month.
- To prepare the bacteria agar-BHI plates, first add antibiotics, if appropriate, to the flask containing BHI and agar (depending on the bacterial strains to be grown). Briefly put the flask on a magnetic stir plate to allow quick mixing.
NOTE: The antibiotics used here are specific to the Lm strains used, but any necessary antibiotics can be used in BHI-agar plates. We added streptomycin to a concentration of 200 µg/mL and chloramphenicol to a concentration of 7.5 µg/mL because 10403S Lm strains are resistant to streptomycin. These strains have been conjugated with a plasmid that contains the chloramphenicol acetyltransferase open reading frame, hence the resistance to chloramphenicol. - Pour the mixture into 10-cm polystyrene bacteria culture plates (approximately 20 mL per plate). To get rid of bubbles, flame the upper surface of the plates briefly. Cool plates overnight at room temperature.
- The next day, seal the dry plates and store them at 4 °C.
- Streptomycin, chloramphenicol, and gentamicin stocks
- Infection of human microvascular endothelial cells with L. monocytogenes
- Three days before the infection, streak out the Lm strain to be used from a glycerol stock (stored at -80 °C) onto a BHI-agar plate that contains 7.5 µg/mL of chloramphenicol and 200 µg/mL of streptomycin, if appropriate.
NOTE: The strain to be streaked out can be a wild-type or mutant, constitutively expressing fluorescence (for immunostaining JAT1045 was used) or expressing fluorescence under the ActA promoter (for flow cytometry or traction force microscopy JAT983 or JAT985 were used)22. - Incubate plates at 37 °C until discrete colonies are formed (1 – 2 days).
- The day before the infection, grow the desired strain overnight, shaking it at 150 rpm at 30 °C in BHI media with 7.5 µg/mL of chloramphenicol (if appropriate).
- Place 5 mL of BHI media in a 15-mL conical centrifuge tube, add 7.5 µg/mL of chloramphenicol (if appropriate), and then inoculate a single colony from the agar plate using a sterile 10 µL tip.
- The next day, just prior to infection, measure the optical density of the bacteria solution at 600 nm (OD600) by diluting the sample 1:5; use a cuvette containing BHI alone to serve as a blank.
- Dilute the overnight culture to an OD600 of 0.1 and incubate it, shaking for 2 h at 30 °C, in BHI media with 7.5 µg/mL of chloramphenicol (if appropriate) to allow the bacteria to reach log-phase growth.
- Measure the OD600 of the bacterial solution, which is expected to be around 0.2 – 0.3. If the OD600 is higher, dilute it to 0.2 – 0.3 with BHI alone.
- Take 1 mL of bacterial solution into a microcentrifuge tube. Spin it down for 4 min at 2,000 x g using centrifugation at room temperature. Remove the supernatant and resuspend the bacterial pellet in 1 mL of tissue culture-grade PBS, in the tissue culture hood. Wash the bacteria twice more by spinning them down for 4 min at 2,000 x g at room temperature. Remove the supernatant and resuspend the bacteria in 1 mL of PBS.
- Prepare the infection mix by mixing 10 or 50 µL of the bacteria resuspended in PBS with 1 mL of MCDB-131 full media for a multiplicity of infection (MOI; i.e., the number of bacteria per host cell) of approximately 50 bacteria per host cell or 10 bacteria per host cell.
- Remove the media from the wells of the 24-well plates, careful not to disrupt the hydrogels or the cells. Wash the cells 1x with 1 mL of MCDB-131 full media and then add 1 mL of the bacteria to each well.
- Keep some infection mix (at least 100 µL) for the determination of MOI (see step 3.3).
- Cover the plate with its lid and wrap the plates with polyethylene food wrap to avoid leakage. Place the plates in the centrifuge and spin the samples for 10 min at 200 x g to synchronize the invasion. Move the plates into the tissue culture incubator and incubate them for 30 min at 37 °C.
- Wash the samples 4x with MCDB-131 full media and move them into the tissue culture incubator. After an additional 30 min, replace the media with media supplemented with 20 µg/mL of gentamicin.
- Three days before the infection, streak out the Lm strain to be used from a glycerol stock (stored at -80 °C) onto a BHI-agar plate that contains 7.5 µg/mL of chloramphenicol and 200 µg/mL of streptomycin, if appropriate.
- Determination of the multiplicity of infection (MOI)
- During the initial 30-min incubation of the host cells with the bacteria, prepare 10-fold serial dilutions of the infection mix to determine the MOI. Make 10-fold dilutions by mixing 100 µL of the infection mix with 900 µL of 1x PBS (10-1 dilution). Then, mix 100 µL of the 10-1 dilution with 900 µL of PBS (10-2 dilution).
- Continue until a 10-5 dilution is obtained.
NOTE: All solutions need to be mixed well before diluting them, and fresh clean pipette tips should be used at each step. - Once the dilutions are made, place 100 µL of the 10-2 – 10-5 dilutions on the center of the BHI/agar/chloramphenicol/streptomycin plates (if appropriate).
- Make a spreader from a glass pipette by using fire to bend the pipette to create a hook. Dip the pipette in 100% ethanol for sterilization and then burn off the ethanol with a flame.
- Once the spreader has cooled, spread the bacterial dilutions homogeneously on the plates starting from the 10-5 dilution and moving to the more concentrated mixes. Incubate the plates upside down at 37 °C for 2 days.
- Depending on the colony density, determine the number of colonies of either the 10-3 and 10-4 or the 10-4 and 10-5 dilution plates. Calculate the MOI as follows:
number of colonies × dilution factor ÷ volume initial added = number of CFU per µL
number of CFU per µL × volume of bacteria mix per well = number of CFU per well
number of CFU per well × volume of cells per well = number of bacteria per cell
NOTE: CFU stands for colony-forming units. - Average the MOIs from two plates to obtain the final MOIs.
4. Flow Cytometry to Quantify Extracellular-matrix-stiffness Dependent Susceptibility of Host Cells to Infection
- Weigh 5 mg of collagenase and place it in a 15-mL conical centrifuge tube. Add 10 mL of 0.25% trypsin-EDTA and mix them well.
- 8 h post-infection, remove the media from the wells of the 24-well plate and wash the wells 1x with tissue culture PBS. Remove the PBS from the wells and add 200 µL of the trypsin-EDTA/collagenase mix to each well. Place this in the tissue culture incubator for 10 min to allow full detachment of the cells.
- Use a fresh pipette for each well and pipet the mix up and down 8x. Be gentle so as to not damage the cells. Add 200 µL of full media to each well to neutralize the trypsin.
- Transfer the 400-µL cell solution of each well into a 5-mL polystyrene tube with a 35-µm cell strainer cap (see Table of Materials).
- Analyze 10,000 – 20,000 cells per sample by flow cytometry. Perform data acquisition through flow cytometry and determine the percentage of infected cells per well using a relevant commercially-available software. Use the forward scatter versus side scatter plots to gate the bulk of the distribution of the cell counts.
NOTE: This will ensure analysis of single cells and eliminate debris or cell doublets or triplets.- Measure the fluorescence signal from control-uninfected cells and gate the population of infected cells excluding autofluorescence.
5. Immunostaining of the Extracellular Bacteria, Microscopy and Image Processing
Note: This approach is followed to differentiate between ECM-stiffness dependent bacterial adhesion onto the host cell surface versus bacterial internalization (invasion) within the hosts.
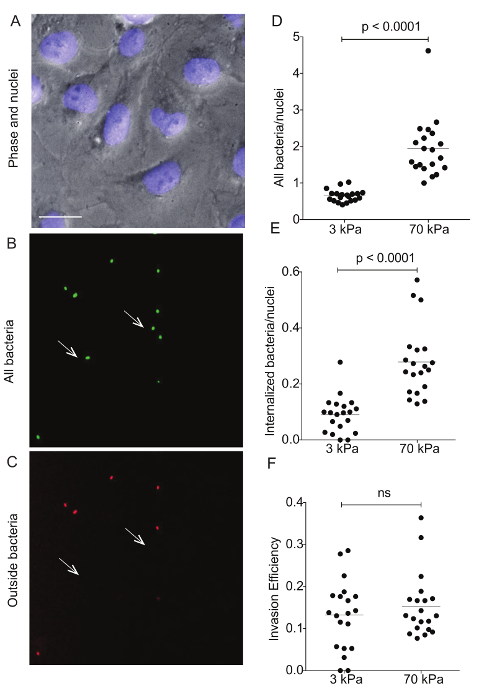
Figure 4: Immunostaining of Lm-infected HMEC-1 cells residing on hydrogels of varying stiffness to differentiate bacterial adhesion versus invasion. These images show a representative example of differential immunostaining showing A. the cell nuclei (DAPI), B. all bacteria (GFP), and C. the outside bacteria (Alexa-546). The HMEC-1 cells were residing on a soft 3-kPa hydrogel. The arrows point at bacteria that have been internalized, so are shown on the green channel only. Data in D–F refer to N = 20 images captured for the infected HMEC-1 cells residing on soft 3-kPa and stiff 70-kPa hydrogels andshow D. the total bacteria per nuclei; E. the internalized bacteria per nuclei;and F. the invasion efficiency (the ratio of internalized bacteria to total bacteria). The horizontal bars depict the data's mean. The P-value was calculated with the non-parametric Wilcoxon Rank Sum test. Please click here to view a larger version of this figure.
- 30 min post-infection, wash the HMEC-1 cells infected with JAT1045 (Lm that constitutively expresses green fluorescent protein (GFP)) 4x with media.
- After the fourth wash, mix 1 µL of 1 mg/mL Hoechst dye with 1 mL of MCDB-131 full media and add it to each well to stain the cells' nuclei. Place the cells in the tissue culture incubator for 10 min.
- Wash the cells 1x with PBS. Fix them with a non-permeabilizing fixative for differential immunostaining for 20 min at room temperature28.
NOTE: The fixative contains 0.32 M sucrose, 10 mM MES pH 6.1, 138 mM KCl, 3 mM MgCl2, 2 mM EGTA, and 4% formaldehyde (electron microscopy grade). - Wash the cells 1x with PBS. Block the samples for 30 min with 5% bovine serum albumin (see Table of Materials) in PBS.
- Incubate the samples for 1 h with an anti-Lmprimary antibody (see Table of Materials) diluted 1:100 in PBS containing 2% BSA.
- Wash the samples in PBS 3x and then incubate them for 1 h with an AlexaFluor-546 goat-anti-rabbit secondary antibody (see Table of Materials) diluted 1:250 in PBS containing 2% BSA. Wash the samples 3x in PBS. Store the samples in 1 mL of PBS for imaging.
- Image and analyze more than 1,000 cells per condition.
- For imaging, use an inverted epifluorescence microscope with a CCD camera (see Table of Materials) and a 40X air plan fluorite air objective with 0.6 numerical aperture (NA).
NOTE: The power is set to 25% and the exposure time to 100 ms. The microscope filters used for mCherry are 470/525 nm, for GFP 530/645 nm, and for DAPI 395/460 nm, for excitation and emission respectively. The microscope we used was controlled by an open source microscopy software package29. - For differential immunostaining, count all 'green' bacteria associated with individual cells as adherent. Count bacteria that are both 'green' and 'red' (due to antibody binding) as non-internalized.
NOTE: We identified the nuclei number by running a custom-made script and the CellC software (see Table of Materials) for enumeration of the bacteria30.
- For imaging, use an inverted epifluorescence microscope with a CCD camera (see Table of Materials) and a 40X air plan fluorite air objective with 0.6 numerical aperture (NA).
6. Multi-well Traction Force Microscopy and Monolayer Stress Microscopy
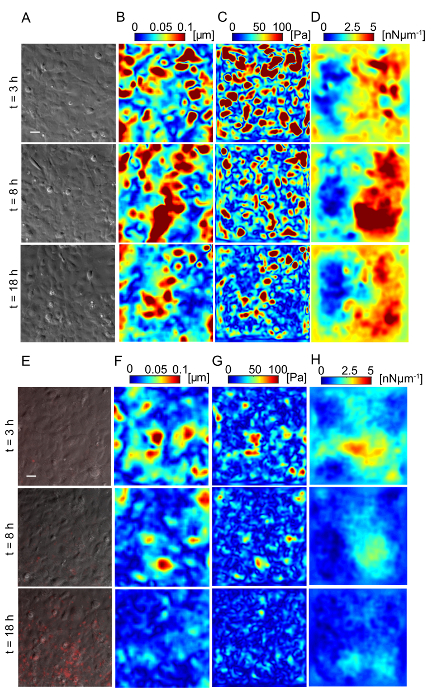
Figure 5: Lm-infected HMEC-1 cells decrease the magnitude of the traction forces they exert on their ECM during infection. Panel A shows the phase image, B shows the deformation field, C shows the traction stress field, and D shows the intracellular tension field of the uninfected HMEC-1 cells residing on a 20-kPa hydrogel. The color bars of the deformation maps (µm), of the traction stress maps (Pa), and of the intracellular tension maps (nNµm-1) are shown on the upper portion of the heat maps. The columns show three representative time points: 3, 8 and 18 h post-infection. E-H. Same as panels A–D but for cells infected with Lm at an MOI of 300. The images of the bacteria (red channel) are superimposed on the phase images of the cells. TFM recordings were conducted by imaging multiple wells simultaneously. The window size for PIV was 32 pixels with an overlap of 16. The scale bar is 32 µm. This figure has been modified from Bastounis and Theriot59. Please click here to view a larger version of this figure.
- Prepare the PA hydrogels on a 24-well plate (see step 1). Seed HMEC-1 cells (see step 2) and infect them with JAT983 as described above (see step 3).
- Prepare a live-microscopy medium by supplementing Leibovitz’s L-15 with 10% fetal bovine serum, 10 ng/mL of epidermal growth factor, and 1 μg/mL hydrocortisone.
NOTE: The L-15 already contains 2 mM L-glutamine, so it is not necessary to add extra as in the case of MCDB-131 media. - 4 h post-infection (when the internalized JAT983 starts fluorescing), mix 1 μL of 1 mg/mL Hoechst dye with 1 mL of L-15 full media and add it to each well to stain the cells’ nuclei. Place the cells in the tissue culture incubator for 10 min and then replace in each well the L-15 full media with 1 mL of L-15 full media supplemented with 20 µg/mL of gentamicin.
- Cover the plate with its lid and place it in a microscope's environmental chamber (see Table of Materials) equilibrated to 37 ˚C.
NOTE: For imaging, use an inverted epifluorescence microscope with a CCD camera (see Table of Materials) and a 40X air plan fluorite air objective with 0.6 NA. - Acquire multi-channel time-lapse images of the fluorescent beads and the fluorescent bacteria, and phase contrast images of the host cells, every 10 min for 4 to 12 h.
NOTE: For the imaging, the power was set to 25% and the exposure time to 100 ms. The microscope filters (excitation/emission) used for mCherry and GFP are 470/525 nm and 530/645 nm respectively. - At the end of the recording, add 500 µL of 10% sodium dodecyl sulfate (SDS) in water in each of the wells.
NOTE: The cells will be detached from the hydrogel and the hydrogel will return to its initial undeformed state since it is elastic. - Take an image of the hydrogel and use it as the reference undeformed image.
- Determine the two-dimensional deformation of the hydrogel at each instant of time using particle image velocimetry (PIV)31, comparing each image of the time-lapse series with the reference image of the undeformed hydrogel. Use the appropriate window sizes and overlap depending on the experiment.
NOTE: We used windows of 32 pixels with an overlap of 16 pixels. - Calculate the 2D traction stresses that cells exert on the hydrogel as described elsewhere32,33.
- Calculate the monolayer tension from the traction stresses as previously described34 by solving the equations of mechanical equilibrium for a thin elastic plate subject to the reaction forces created by the PA hydrogel on the monolayer, which are of an opposite sign to the measured traction stresses.
NOTE: See Supplementary Material 1.
7. Quantitative Time-lapse Microscopy to Assess Extracellular-matrix-stiffness Dependent L. monocytogenes Dissemination Through Endothelial Cells
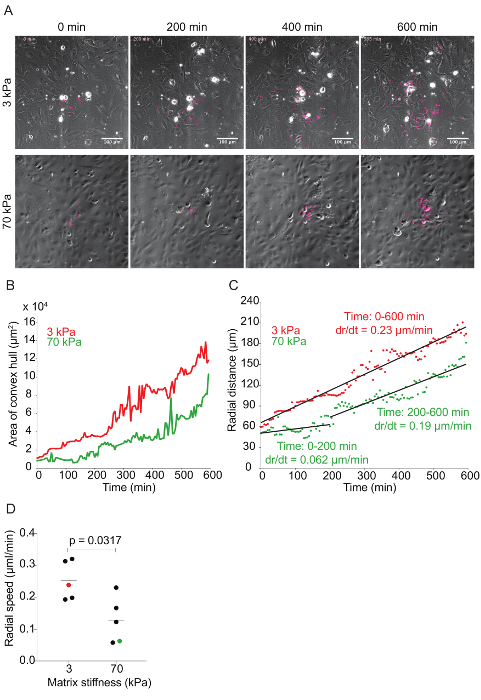
Figure 6: Time-lapse quantitative microscopy data of Lm dissemination through HMEC-1 monolayers seeded on substrates with 3-kPa and 70-kPa stiffness. A. This panel shows still images from two representative infection foci at 0, 200, 400, and 600 min. The phase channel depicts HMEC-1 cell monolayers, and the red channel depicts intracellular Lm (see step 7 of the protocol). B. This panel shows the area of the convex hull encompassing the infection focus plotted as a function of time. The focus area of the 3-kPa condition (red) grows faster than that of the 70-kPa (green). C. This panel shows the radial distance from the center to the edge of the infection focus plotted as a function of time for representative infection foci growing in HMEC-1 monolayers seeded on PA substrates of 3-kPa (red) and 70-kPa (green) stiffness. The radial speed (dr/dt) is constant for the 3-kPa condition, but biphasic for the 70-kPa condition. D. These data show the radial speed for the first 200 min of ten independent infection foci. The red (3-kPa) and green (70-kPa) data points depict the slopes of the two foci described in the previous panels. The horizontal bars depict the data's mean. The P-value was calculated with the non-parametric Wilcoxon Rank Sum test. Please click here to view a larger version of this figure.
- Prepare the PA hydrogels on a 24-well plate (see step 1), seed HMEC-1 cells (see step 2), and grow overnight JAT983 cultures as described above (see step 3).
- The day of infection, prepare the infection mix by mixing 10 µL of bacterial pellet per mL of MCDB-131 full media. Carefully remove the media from the wells of the 24-well plates, and add 1.9 mL of the MCDB-131 full media and 0.1 mL of the bacterial mix to each well.
NOTE: The lower MOI used in this assay is necessary to analyze dissemination events that start from a single bacterium invading a host cell. - Cover the plate with its lid and seal it with a plastic wrap to avoid leakage. Place the plates in the centrifuge and spin the samples for 10 min at 200 x g to synchronize the invasion.
- Move the plates into the tissue culture incubator and incubate them for 5 min.
- Wash the samples 4x with media and move them to the tissue culture incubator. After an additional 5 – 7 min, replace the media with media supplemented with 20 µg/mL of gentamicin.
- Incubate the plate in the incubator for an extra 5 h to allow the actA promoter to turn on and drive the expression of the mTagRFP open reading frame35.
- Prepare a live-microscopy medium by supplementing Leibovitz's L-15 with 10% fetal bovine serum, 10 ng/mL of epidermal growth factor, and 1 µg/mL hydrocortisone.
NOTE: The L-15 already contains 2 mM L-glutamine, so it is not necessary to add extra as in the case of MCDB-131 media. - Mix 1 μL of 1 mg/mL Hoechst dye with 1 mL of L-15 full media and add it to each well to stain the cells’ nuclei. Place the cells in the tissue culture incubator for 10 min and then replace in each well the L-15 full media with 1 mL of L-15 full media supplemented with 20 µg/mL of gentamicin. Cover the plate with its lid and place it in a microscope’s environmental chamber (see Table of Materials) equilibrated to 37 ˚C.
NOTE: For imaging, an inverted epifluorescence microscope was used with a CCD camera (see Table of Materials) and a 20X air plan fluorite air objective with 0.75 NA. The power was set to 50% and the exposure time to 50 ms. The microscope filters used for mCherry are 470/525 nm for excitation and emission respectively. - Image multiple positions every 5 min using an autofocus feature to monitor how Lm spread through HMEC-1 monolayers seeded on varying stiffness hydrogels.
NOTE: L-15 is buffered with HEPES, so it is not necessary to use CO2 during the imaging.
ECM stiffness-dependent susceptibility of HMEC-1 cells to Lm infection:
PA hydrogels of varying stiffness, all surface-coated with collagen I, were built on multi-well glass bottom plates as described in step 1 of the protocol (see Figure 1). AFM measurements were performed to confirm the exact stiffness of the hydrogels, as described previously26,27 (see Figure 2). Past studies have shown that the local compliance of the basement membrane of endothelial cells can range from 1 kPa (e.g., brain tissue) to 70 kPa (e.g., aorta)36,37,38,39,40. Therefore, we chose to test the infection of HMEC-1 cells residing on matrices with the following stiffness: 0.6 kPa, 3 kPa, 20 kPa, and 70 kPa. For each hydrogel stiffness, six hydrogels were fabricated to assess the reproducibility of the results.
Flow cytometry was used to assess the ECM stiffness-dependent susceptibility of HMEC-1 cells to Lm infection (see Figure 3). HMEC-1 cells were infected with a Lm strain (JAT985) that expresses a fluorescent marker after internalization (actAp::mTagRFP), allowing the detection of intracellular bacteria only (see Figures 1L – 1N). JAT985 also lacks ActA, disabling the bacteria from spreading from cell to cell, since ActA is necessary for the formation of actin comet tails and subsequent bacterial dissemination. 7 – 8 h post-infection, the HMEC-1 cells' infection was assessed using flow cytometry. To ensure the analysis of single cells, the bulk of the distribution of cell counts was gated using the forward versus side scatter plot, and then a second gating step was performed to exclude cells that exhibit autofluorescence (see Figures 3A – 3C). The preliminary results depicted in Figure 3 show that HMEC-1 infection with Lm is approximately two-fold greater for HMEC-1 cells residing on the stiff 70 kPa hydrogels as compared to cells residing on the soft 0.6 kPa hydrogels (see Figures 3B – 3D). The increased Lm infection susceptibility of HMEC-1 cells residing on stiff as compared to soft matrices could be due to: 1. increased Lm adhesion onto HMEC-1; 2. increased Lm invasion into HMEC-1; or 3. both of the above co-occurring. To test which hypothesis holds, HMEC-1 cells were seeded on soft (3 kPa) and stiff (70 kPa) hydrogels and infected with constitutively GFP expressing Lm (see Figures 4A – 4C). The samples were fixed shortly after infection and the external (adhering) bacteria were stained with antibodies. Using quantitative microscopy, we found that there are significantly more bacteria adhering to HMEC-1 when the host cells reside on stiff as compared to soft gels (see Figure 4D). Consistent with the flow cytometry data, there are significantly more bacteria internalized by HMEC-1 when the host cells reside on stiff as compared to soft gels (see Figure 4E). However, the invasion efficiency (bacteria internalized/total number of bacteria) of Lm into HMEC-1 cells is similar, irrespective of substrate stiffness (see Figure 4F).
Traction force microscopy of Lm-infected HMEC-1 cells:
Lm infection of HMEC-1 cells could alter the biomechanics of infected host cells, including the strength of attachment to their ECM or to each other, affecting their barrier integrity. We sought to evaluate whether that could be the case by using Traction Force Microscopy32 to calculate the cell-ECM traction forces and Monolayer Stress Microscopy41 to calculate the intracellular tensional forces. Figure 5 depicts maps of the deformation field, traction stress field, and intracellular tension field of HMEC-1 cells residing on 20-kPa hydrogels at different time points post-infection. Figures 5A – 5D refer to uninfected control HMEC-1 cells while Figures 5E – 5H refer to HMEC-1 cells infected with Lm at a multiplicity of infection equal to 300 bacteria/cell. This preliminary work suggests that infected HMEC-1 cells reduce the magnitude of their cell-ECM and intracellular stresses during the course of an infection with Lm, whereas that is not observed for uninfected control cells.
ECM stiffness-dependent Lm dissemination across HMEC-1 monolayers:
Time-lapse microscopy was used to investigate the effect matrix stiffness has on Lm dissemination through HMEC-1 monolayers. As Lm spreads through the monolayer, the bacteria create a focus of infection that grows as a function of time (see Figure 6A and Video Figures 1 and 2). The area of the infection focus was measured by drawing a convex hull, the smallest convex polygon that encompasses a set of points, around the bacteria42. There is no standard metric in the field to measure the efficiency of L. monocytogenes cell-to-cell spread through a monolayer of host cells. To assess spread efficiency, some have counted the number of host cells in an infection focus41, and others have drawn boundaries manually around the group of infection host cells43. We chose to draw a convex polygon around the bacteria, because it is an automated, consistent, and computationally inexpensive process to measure the efficiency of L. monocytogenes spread. By doing so, we found a slight decrease in the infection focus area in the HMEC-1 cells seeded onto 70 kPa hydrogels when compared to those seeded on 3 kPa matrices (see Figure 6B and Video Figures 1 and 2). To determine the rate of growth of the infection focus, the radial distance was plotted as a function of time by taking the square root of the area of the infection focus and dividing this by pi. This mathematical transformation assumes that the shape of the infection focus is roughly circular44. To measure the speed of the focus growth, the rate of change (i.e., the slope) of the radial distance for both 3- and 70-kPa matrices was measured. This approach elucidated that the infection focus grew faster and monotonically in HMEC-1 cells seeded onto 3-kPa matrices. However, the focus grew significantly slower (first 200 min) and slightly slower (200 to 600 min) in cells seeded onto 70-kPa matrices (see Figure 6C). Indeed, analysis of further data confirmed that the infection focus grew, on average, two-fold slower in 70-kPa matrices, especially during the first 200 min (see Figure 6D).
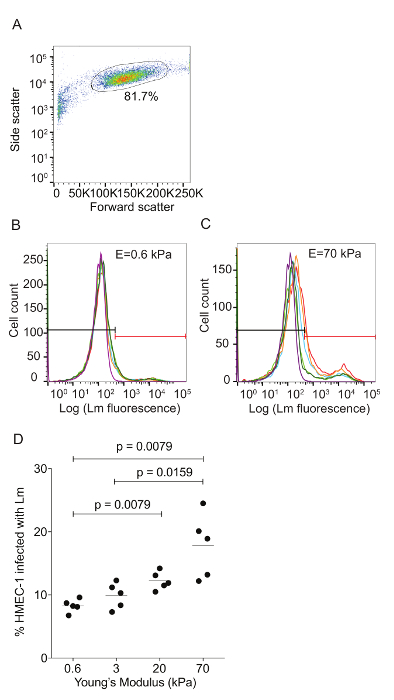
Figure 3: ECM stiffness-dependent susceptibility of HMEC-1 cells to Lm invasion measured by using flow cytometry. HMEC-1 cells were infected with Lm (JAT985) and the infection was analyzed by flow cytometry. A. This panel shows a side versus forward scatter plot for representative HMEC-1 cells coming from a single well. The bulk distribution of cells was selected via gating to exclude debris (left) and cell doublets or triplets (right). B. This graph shows a histogram of the logarithm of the Lm fluorescence intensity per cell for HMEC-1 plated on soft 0.6 kPa. C. This graph shows a histogram of the logarithm of the Lm fluorescence intensity per cell for HMEC-1 plated on stiff 70 kPa hydrogels. The histograms for N = 4 – 6 replicates are shown in different colors. The control uninfected cells' histogram is purple. The gate used to define what is infected is shown in red. The MOI is 100 and the infection was assessed 8 h post-infection. D. Data show percentage of infected HMEC-1 cells versus hydrogel stiffness (N = 5). The horizontal bars depict the data's mean. The P-value was calculated with the non-parametric Wilcoxon Rank Sum test. Please click here to view a larger version of this figure.
Video Figure 1: Dissemination of intracellular Lm (red channel) through an HMEC-1 cell monolayer (phase) seeded on a 3-kPa substrate. The cells were imaged in Leibovitz's L-15 media (10% FBS, 20 µg/mL of gentamicin) inside an environmental chamber equilibrated to 37 ˚C. The images were collected every 5 min. The movie speed is 15 frames/s. Please click here to view this video. (Right-click to download.)

Video Figure 2: Dissemination of intracellular Lm (red channel) through an HMEC-1 cell monolayer (phase) seeded on a 70-kPa substrate. The cells were imaged in Leibovitz's L-15 media (10% FBS, 20 µg/mL of gentamicin) inside an environmental chamber equilibrated to 37 °C. The images were collected every 5 min. The movie speed is 15 frames/s. Please click here to view this video. (Right-click to download.)
| Young’s modulus (E, kPa) | Acrylamide % (from 40% stock) | Bisacrylamide % (from 2% stock) |
| 0.6 | 3 | 0.045 |
| 3 | 5 | 0.075 |
| 10 | 10 | 0.075 |
| 20 | 8 | 0.195 |
| 70 | 10 | 0.45 |
Table 1. Composition of polyacrylamide (PA) hydrogels of varying stiffness. In this table, the percentage of stock 40% acrylamide solution and the percentage of stock 2% bis-acrylamide solution to achieve a given stiffness (Young's modulus, E) are indicated in different columns.

Supplemental Material 1. Computation of intracellular tension from calculated traction stresses. Please click here to download this file.
