Induction of differentiation and generation of swarming colonies
Figure 1 provides a schema of the important steps involved in producing swarming colonies of V. parahaemolyticus (Figure 1A-C). A swimmer culture was spotted on swarm-agar and incubated at 24 oC, inducing swarmer differentiation and proliferation over the solid agar surface. A representative stereo-microscopy image of a swarming colony is presented in Figure 2. Stereo-microscopy imaging clearly shows swarm-flares extending outward of the swarm-colony periphery. Higher magnification reveals that in swarm-flares cells are grouped in mono- or bi-layers only, whereas in the middle of the swarm-colony cells are stacked in multiple layers (Figure 2A). Swarm-flares can easily be transferred (as described in steps 3.8-3.11, Figure 1) to a microscope slide. Importantly, the flares only consist of a fully differentiated population of swarmer cells (Figure 2B). This is in contrast to cells from the middle of the swarm-colony, which are significantly shorter and likely represent a mixture of swimmer and swarmer cells (Figure 2B)9. Furthermore, careful transfer of the flare to the microscope slide leaves the overall structure and swarm-edge of the swarm-flare intact, resulting in a mono-layer of swarmer cells accessible for single cell microscopy. When imprinting the swarm-flares on the microscope slide, it is very important to do the transfer as gentle as possible and not to add too much pressure – otherwise cells from swarm-flares will spread over the slide mixing with non-swarmer cells originating from the middle of the swarm-colony.
Fluorescence microscopy and image analysis
As proof of principle of single cell microscopy of differentially distinct V. parahaemolyticus swimmer and swarmer cells, a detailed analysis of ectopically expressed YFP-CheW was performed. CheW is an essential chemotaxis protein and can be used as a marker for the intracellular localization of chemotactic signaling arrays9,12. A plasmid encoding YFP-CheW under an L-arabinose inducible promoter was electroporated into V. parahaemolyticus as described in section 1 of the protocol. For microscopy, swimmer and swarmer cells were prepared as described in sections 4 and 3 respectively. As previously described YFP-CheW was localized in a uni-polar (Figure 3A, red arrows) and bi-polar manner (Figure 3A, yellow arrows)12,13. Accordingly, identification of the cell pole and the variation of YFP-protein localization at different lengths offer a great insight in how the localization pattern evolves as the cell cycle proceeds. These data can then be displayed in scatter plots correlating the variables "cell length" and "distance of the foci to the cell pole" (Figure 3B). To avoid imprecisions in contour detection, which is common when using automatic cell detection tools, cells are selected by means of manually drawing a line from one pole to the other using the "Multi-line" option in MetaMorph or "Segmented line" in the open-source platform ImageJ. Subsequently, the intensity profiles of the selected region of interest (ROI) can be collected, allowing the identification of the point in which YFP-protein displayed its highest intensity and also the overall length of the cell. The fluorescence profiles of single cells were analyzed as described in section 5 and were presented by plotting the distance of foci to the cell poles as a function of cell length (Figure 3B). This analysis clearly shows that short cells only display a uni-polar localization pattern of YFP-CheW, whereas in longer cells YFP-CheW is bi-polarly localized. Furthermore, localization patterns over the cell cycle can be easily visualized in a demographic representation of the cell length and the fluorescent intensities along the cell body. These representations can be obtained using the same ROIs previously selected when preparing the scatter plots. Hence, a demographic analysis of the whole cellular fluorescence profile as described in step 5.9 (Figure 3C) was performed. This analysis clearly shows a similar pattern of YFP-CheW localization, with YFP-CheW being uni-polarly localized in short cells and bi-polarly localized in longer cells. Thus, indicating a cell cycle-dependent localization pattern, where YFP-CheW is localized uni-polarly in recently divided cells (short), and then gets recruited to the opposite pole later in the cell cycle (long cells), resulting in a bi-polar localization. An analogous analysis was performed on swarmer cells (Figure 4). A population based analysis shows major differences in the localization of YFP-CheW in swarmer cells when compared to that of the swimmer cell-type. YFP-CheW is always bi-polarly localized (Figure 4A, yellow arrows) and importantly YFP-CheW also forms clusters positioned randomly along the cell length (Figure 4A, orange arrows). Thus, there are major changes in the intracellular localization of chemotaxis signaling arrays during differentiation of V. parahaemolyticus. This example shows that the method described allows for the production of a swarmer cell-population that is easily available for single cell fluorescence microscopy. Furthermore, it shows that the pipeline described for image analysis allows for demographic analysis of the intracellular organization of V. parahaemolyticus swarmer cells.
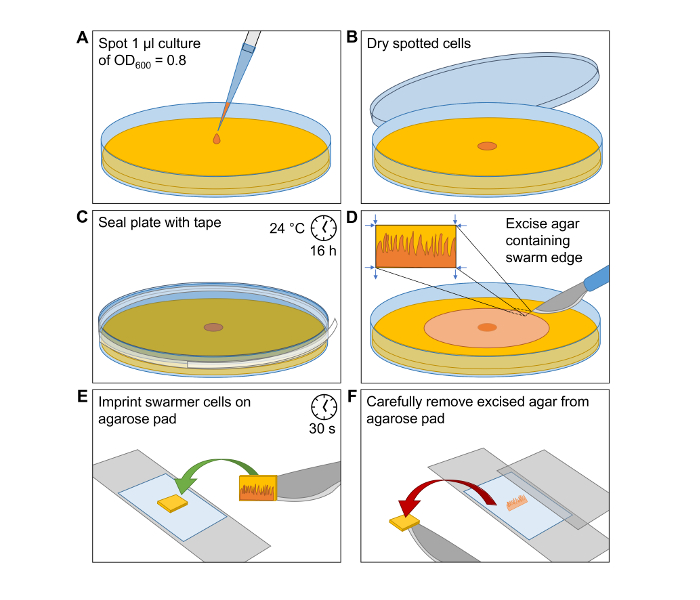
Figure 1: Induction of Swarmer Differentiation and Preparation of Cells for Microscopy Imaging. Simplified visualization of the workflow to produce V. parahaemolyticus swarmer cells and the preparation of microscope slides for microscopy analysis of single swarmer cells. (A) Spot 1 µL of an OD600 = 0.8 cell culture of V. parahaemolyticus at the center of a HI agar swarming plate. (B) Incubate the spot until it is dried and cells are attached to the agar surface. (C) Seal the Petri dish with clear tape. Incubate the plate at 24 °C for 16 h. (D) Carefully excise a small piece of HI agar from the outer edge of the swarming colony. (E) Transfer the excision onto a microscopy agarose pad with the swarmer cells facing the agarose pad. After a short incubation time of 30 s the swarmer cells are imprinted onto the agarose pad. (F) Carefully remove excised agar from agarose pad and place a microscopy coverslip on top. Please click here to view a larger version of this figure.
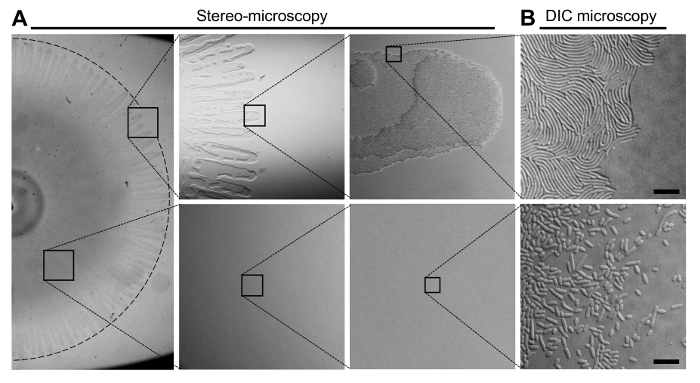
Figure 2: The Organization of Cells in a V. parahaemolyticus Swarming Colony. (A) Stereo-microscopy of a V. parahaemolyticus swarmer colony. The upper panels display the outer edge of a swarming colony in increasing magnification. The bottom panels display an area of the swarming colony between the center and the edge in increasing magnification. Swarm-flares extend from the periphery of the swarm-colony. (B) Representative DIC microscopy images of the swarmer colony edge (upper panel) and an area between the center and the periphery (bottom panel) after transfer of cells from the respective indicated locations of the swarming colony. The swarm-flares were imprinted directly onto a microscopy agarose pad as described in steps 3.8-3.11. Cells from the area between the center and the periphery were first scraped off the swarm-colony and re-suspended in LB broth, before being spotted onto a microscopy agarose pad. Scale bar = 7.5 µm. Please click here to view a larger version of this figure.
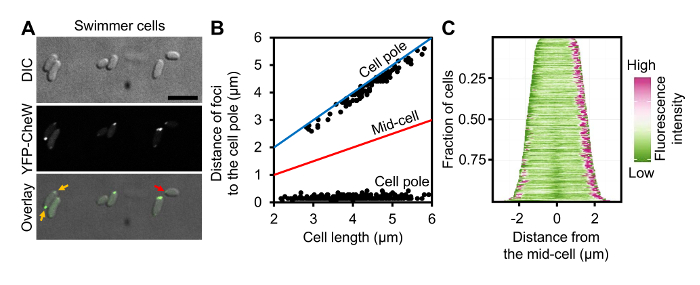
Figure 3: Localization of Chemotaxis Arrays in Swimmer Cells of V. parahaemolyticus. (A) DIC and fluorescent microscopy images of wild-type V. parahaemolyticus swimmer cells ectopically expressing YFP-CheW. (B) Graph displaying YFP-CheW foci distribution by plotting the distance of foci from the cell poles as a function of cell length in swimmer cells. (C) Demographic analysis of fluorescence intensity profiles of V. parahaemolyticus swimmer cells expressing YFP-CheW. Cells are sorted by cell length and displaying relative fluorescent intensity. Scale bar = 5 µm. Panel B and C are adapted from reference9. Please click here to view a larger version of this figure.
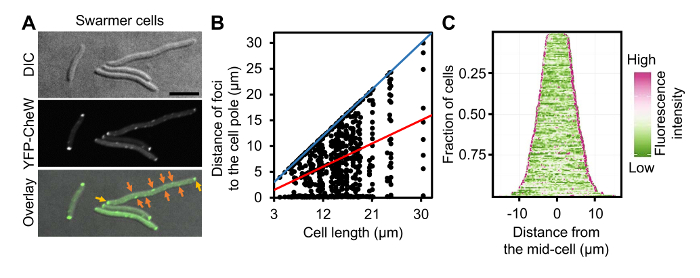
Figure 4: Localization of Chemotaxis Arrays in Swarmer Cells of V. parahaemolyticus. (A) DIC and fluorescent microscopy images of wild-type V. parahaemolyticus swarmer cells ectopically expressing YFP-CheW. (B) Graph displaying YFP-CheW foci distribution by plotting the distance of foci from the cell poles as a function of cell length in swarmer cells. (C) Demographic analysis of fluorescence intensity profiles of V. parahaemolyticus swarmer cells expressing YFP-CheW. Cells are sorted by cell length and display relative fluorescent intensity. Scale bar = 5 µm. Panel B and C are adapted fromreference9. Please click here to view a larger version of this figure.
