In Vitro Colony Assays for Characterizing Tri-potent Progenitor Cells Isolated from the Adult Murine Pancreas
概要
In vitro colony assays to detect self-renewal and differentiation of progenitor cells isolated from adult murine pancreas are devised. In these assays, pancreatic progenitors give rise to cell colonies in 3-dimensional space in methylcellulose-containing semi-solid medium. Protocols for handling single cells and characterization of individual colonies are described.
Abstract
Stem and progenitor cells from the adult pancreas could be a potential source of therapeutic beta-like cells for treating patients with type 1 diabetes. However, it is still unknown whether stem and progenitor cells exist in the adult pancreas. Research strategies using cre-lox lineage-tracing in adult mice have yielded results that either support or refute the idea that beta cells can be generated from the ducts, the presumed location where adult pancreatic progenitors may reside. These in vivo cre-lox lineage-tracing methods, however, cannot answer the questions of self-renewal and multi-lineage differentiation-two criteria necessary to define a stem cell. To begin addressing this technical gap, we devised 3-dimensional colony assays for pancreatic progenitors. Soon after our initial publication, other laboratories independently developed a similar, but not identical, method called the organoid assay. Compared to the organoid assay, our method employs methylcellulose, which forms viscous solutions that allow the inclusion of extracellular matrix proteins at low concentrations. The methylcellulose-containing assays permit easier detection and analyses of progenitor cells at the single-cell level, which are critical when progenitors constitute a small sub-population, as is the case for many adult organ stem cells. Together, results from several laboratories demonstrate in vitro self-renewal and multi-lineage differentiation of pancreatic progenitor-like cells from mice. The current protocols describe two methylcellulose-based colony assays to characterize mouse pancreatic progenitors; one contains a commercial preparation of murine extracellular matrix proteins and the other an artificial extracellular matrix protein known as a laminin hydrogel. The techniques shown here are 1) dissociation of the pancreas and sorting of CD133+Sox9/EGFP+ ductal cells from adult mice, 2) single cell manipulation of the sorted cells, 3) single colony analyses using microfluidic qRT-PCR and whole-mount immunostaining, and 4) dissociation of primary colonies into single-cell suspensions and re-plating into secondary colony assays to assess self-renewal or differentiation.
Introduction
The pancreas is composed of three major cell lineages; acinar cells secrete digestive enzymes, ducts secrete mucin to fend off pathogens and transport digestive enzymes to the gut, and endocrine cells secrete hormones, including insulin and glucagon, that maintain glucose homeostasis. During the embryonic development of the pancreas, the early ductal cells are the source of the tri-potent progenitor cells capable of giving rise to the three lineages in the pancreata of adult animals 1,2. Because adult stem and progenitor cells, such as bone marrow stem cells, are already successfully used to treat various diseases 3, there is intense interest in finding the stem and progenitor cells in the adult pancreas. If isolation and manipulation of adult pancreatic stem and progenitor cells were possible, these cells could be used to treat diseases such as type 1 diabetes, in which the insulin-secreting cells are destroyed by autoimmunity.
Whether tri-potent progenitor cells still exist in adult pancreatic ducts after the completion of embryonic development is a question that is heavily debated in the scientific community. In this debate, and using in vivo cre-lox lineage-tracing techniques, Inada and coworkers showed that adult murine ductal cells labeled with a marker, carbonic anhydrase II, could give rise to all three pancreatic lineages 4. However, using other ductal markers, such as HNF1b 5 and Sox9 2, it was concluded that ductal cells are not the major source of beta cells in adult mice.
Several years ago, we proposed that the cause of the aforementioned debate may be due to the lack, in the field 6,7, of appropriate analytical tools that can be used to measure self-renewal and multi-lineage differentiation-two criteria necessary to define a stem cell. The in vivo cre-lox lineage-tracing technique mentioned above can provide evidence for the progenitor-progeny relationship on a population level. However, this lineage tracing technique is limited in its power to discern whether single progenitor cells can self-renew and differentiate into multiple lineages. Single-cell analysis is important because if several mono-potent progenitors, each with a different lineage potential, were analyzed together, they may collectively appear to have multi-lineage differentiation abilities. In addition, stem cells are usually a minor population of an adult organ. The activities of a minor cell population could be masked by the major population. Therefore, a negative result from a population study does not necessarily indicate the absence of stem cells. Finally, cre-lox lineage tracing does not currently allow the measurement of self-renewal.
To begin addressing the technical gap in the field of pancreatic progenitor cell biology, colony 7-11 or organoid 12-15 assays using 3D culture systems were devised. Two colony assays for pancreatic progenitors were developed in our laboratory: one contains a commercial preparation of murine extracellular matrix proteins (ECM) (see Methods and Equipment Table), and the other contains laminin hydrogel, a defined artificial ECM protein 7-11. Progenitor cells are mixed in semi-solid medium containing methylcellulose. Methylcellulose is a biologically inert and viscous material prepared from wood fibers, and has been routinely used in hematopoietic colony assays 16. The methylcellulose-containing semi-solid medium restricts the movement of single progenitor cells so that they cannot re-aggregate. Yet, the medium is soft enough to allow a progenitor cell to grow and differentiate into a colony of cells in the 3D space. Following the tradition of the hematologists, a pancreatic progenitor cell that was capable of giving rise to a colony of cells was named a pancreatic colony-forming unit (PCFU). PCFUs, when grown in the murine ECM-containing colony assay, give rise to cystic colonies that are named "Ring" colonies 7. Upon addition of a Wnt agonist, R-spondin1, into the murine ECM-containing culture, some Ring colonies turn into "Dense" colonies 7. In this article, these two types of colonies grown in murine ECM culture are collectively referred to as "Ring/Dense" colonies. When Ring/Dense colonies are dissociated into single cell suspension and re-plated into cultures that contain laminin hydrogel, "Endocrine/Acinar" colonies are formed 7.
Using single colony analyses, it was found that the majority of Ring/Dense and Endocrine/Acinar colonies, either from adult (2-4 month-old) 7,11 or young (1 week-old) 9 murine pancreas, express all three lineage markers. This suggests that most of the originating PCFUs are tri-potent. In the murine ECM-containing colony assay, adult murine PCFUs robustly self-renew and expand approximately 500,000 times over 11 weeks in culture 7. Murine ECM preferentially supports the differentiation of ductal cells over endocrine and acinar lineages, whereas in the presence of laminin hydrogel, murine PCFUs are encouraged to differentiate preferentially into endocrine and acinar cells and less so to the ductal lineage 7,9,11. Importantly, Insulin+Glucagon– mono-hormonal cells are generated in the laminin hydrogel culture and secrete insulin in response to glucose stimulation in vitro 7,9, suggesting functional maturity. The tri-lineage differentiation potential 7,9 and self-renewal 11 of individual PCFUs are confirmed by single-cell micromanipulation, i.e., culturing one cell per well for colony formation. Together, these results provide evidence that there are self-renewing, tri-potent, progenitor-like cells in the postnatal murine pancreas that show activities in 3D culture.
The murine PCFU assays described in this article are derived from a prior colony assay designed for progenitor cells differentiated from murine embryonic stem cells (mESCs) 17. That protocol is documented in detail in another JoVE publication 18. The culture components and techniques required to perform the murine ECM-containing colony assay for adult PCFUs are the same as for mESC-derived progenitors 17,18. Therefore, these aspects of the assay will not be repeated here; instead the following procedures will be addressed: 1) dissociation of the adult pancreas and sorting CD133+Sox9/EGFP+ ductal cells, which enrich PCFUs from adult mice 7, 2) single-cell manipulation of the sorted cells, 3) single-colony analyses using microfluidic qRT-PCR and whole-mount immunostaining, and 4) dissociation of colonies into single-cell suspension and re-plating into murine ECM or laminin hydrogel colony assays.
Protocol
Ethical Statement: We adhere to the widely accepted ethical standards in conducting research to ensure the quality and the integrity of the results. Animal experimentation is conducted according to protocols approved by the Institutional Animal Care and Use Committee at City of Hope.
1. Prepare Single-cell Suspension from Adult Murine Pancreata
NOTE: In prior publications 7,11, CD-1 or B6 background mice were used; both backgrounds yielded similar results. A transgenic mouse line (designated Sox9/EGFP) with enhanced green fluorescence protein driven by Sox9 loci 19,20, was created in the CD-1 background.
- Euthanize 3 to 5 adult mice using gas CO2 for 1-2 min or until breathing stops. Next, perform cervical dislocation on each mouse.
- Dissect and Prepare the Pancreata.
NOTE: Dissect the mice as soon as possible after euthanasia. This is important to avoid auto-digestion of the pancreas due to post-mortem changes.- Make a vertical incision on the midline of the abdominal wall of the mouse using scissors and open the abdominal cavity. Find the spleen and use forceps to gently lift it. Cut the connective tissue between the spleen and the splenic lobe of the pancreas with scissors.
- Repeat this practice for the duodenal and gastric lobes of the pancreas. Place the pancreatic tissues in a Petri dish on ice containing cold Dulbecco's modified phosphate buffer solution (DPBS), 0.1% bovine serum albumin (BSA), and penicillin and streptomycin (P/S; complete solution designated as PBS/BSA).
- Remove fat tissues from the pancreas under a dissecting microscope using fine-tip forceps.
NOTE: This is critical for the health of the dissociated pancreatic cells in the following steps.- (Optional) Check the pancreata for green fluorescence under the fluorescence stereomicroscope using 488-509 nm excitation to ensure EGFP expression.
- In a tissue culture hood, rinse the tissue three times sequentially in three 100 mm Petri dishes containing 10 ml of cold PBS/BSA.
- Make a vertical incision on the midline of the abdominal wall of the mouse using scissors and open the abdominal cavity. Find the spleen and use forceps to gently lift it. Cut the connective tissue between the spleen and the splenic lobe of the pancreas with scissors.
- Generate Small Pieces of Tissues.
- Transfer the dissected pancreata to a dry sterile Petri dish to remove as much PBS/BSA as possible, and transfer them to another dry Petri dish on ice.
- Prepare PBS/BSA containing DNase I by adding 2 µl DNase I stock solution (1 million units [MU] /ml) per 1 ml PBS/BSA.
NOTE: This solution is designated PBS/BSA/DNase I. Make ~200 ml of this solution at once, which will cover one sorting experiment. - Mince the pancreata using spring scissors for 2-3 min or until the tissue is in fine pieces. Add 2-3 ml of cold PBS/BSA/DNase I to the tissue pieces in the Petri dish to suspend them.
- Transfer the tissue pieces to a 50 ml conical tube on ice with a 10 ml pipette. Bring the total volume to 10 ml with PBS/BSA/DNase l after recovering as much tissue as possible.
- Digest the Pancreatic Pieces into Mostly Single Cells.
- Add collagenase B (100 mg/ml stock) at 350-450 µl/10 ml to the tissue pieces and incubate the tissue at 37 °C in a water bath for 8 min, swirling every 3-4 min. Use a 10 ml syringe with a 16 ½ G needle to draw up and subsequently spray the tissue solution down the wall of the 50 ml tube at RT. Repeat this seven times.
NOTE: Use enough force to break up the cell clusters but avoid generating bubbles that may kill the cells. - Return the tube to the water bath at 37 °C for 8 min and mix by swirling every 3-4 min. Syringe the tissue up and down seven times, as mentioned above, and place a sample (10 µl) of the tissue solution on a dry Petri dish. Observe the tissue solution under an inverted, phase-contrast, light microscope with a 10X objective lens. Expect single cells and some small-sized cell clusters to be present.
- Handle the cells on ice from this point on to slow or stop the activity of collagenase. Adjust the volume to 50 ml using cold PBS/BSA/DNase I. Centrifuge the cells at 400 x g for 5 min at 4 °C, and resuspend in 5 ml of cold PBS/BSA/DNase I using a P1000 pipettor.
- Add collagenase B (100 mg/ml stock) at 350-450 µl/10 ml to the tissue pieces and incubate the tissue at 37 °C in a water bath for 8 min, swirling every 3-4 min. Use a 10 ml syringe with a 16 ½ G needle to draw up and subsequently spray the tissue solution down the wall of the 50 ml tube at RT. Repeat this seven times.
- Filter to Yield Single Cell Suspensions and Wash.
- Filter the cells through a 70 µm nylon mesh filter cup and then through a 40 µm nylon mesh filter cup.
- Resuspend the cells in 5 ml cold PBS/BSA/DNase I and count the cells.
NOTE: For 2-4 month-old B6 or CD-1 mice, each pancreas may yield ~5 or 10 million cells, respectively. These numbers may vary among different experimentalists. If excessive cell debris is found at counting, bring the volume to 50 ml with cold PBS/BSA/DNase I and centrifuge the cells at 400 x g for 5 min at 4 °C. - Adjust the volume of the cell suspension to a concentration of 2 x 107 cells/ml using cold PBS/BSA/DNase I.
2. Sort the Cells to Enrich Pancreatic Colony-forming Progenitors
NOTE: From B6 mice, CD133+ but not CD133– cells are enriched for PCFUs 7,9,11. CD133+ cells represent ~13% of the total dissociated pancreatic cells after all gating parameters are applied11. From CD-1 mice, CD133+Sox9-EGFP+ cells typically represent ~4% of total pancreatic cells, and this cell population is enriched for PCFUs 7. Sorting of the CD133+Sox9/EGFP+ cells is described here.
- Stain the Dissociated Pancreatic Cells.
- Block the entire pancreatic single-cell suspension to reduce non-specific binding by incubating the single-cell suspension with anti-mouse CD16/32 at 10 µg/ml final concentration for 5 min on ice.
- Remove an aliquot of 1 x 106 cells (50 µl) to stain with the isotype control antibody, biotin-conjugated rat IgG1 at 5 µg/ml final concentration, for 20 min on ice, swirling every 5-7 min.
- Remove an aliquot of 1 x 106 cells (50 µl) and keep on ice as an unstained control.
- Stain the remainder of the cells with a biotin-conjugated anti-mouse CD133 primary antibody, at 5 µg/ml final concentration, for 20 min on ice, swirling every 5-7 min.
- Washing Cells.
- Wash the isotype control and primary antibody-stained samples by bringing the volume to 1 ml with PBS/BSA/DNase I and centrifuge at 400 x g for 5 min at 4 °C. Repeat this washing step.
- Resuspend the isotype control and primary antibody-stained samples in PBS/BSA/DNase I, so the final concentration is 2 x 107 cells/ml.
- Stain the isotype control and primary antibody-stained samples with streptavidin (STV)-labeled allophycocyanin (APC) at 2 µg/ml final concentration. Incubate for 15 min on ice, swirling every 5-7 min.
- Washing cells and 4',6-diamidino-2-phenylindole (DAPI) staining.
- Wash the isotype control and primary antibody-stained samples twice with PBS/BSA/DNase I, as in step 2.2. Resuspend the cells in PBS/BSA/DNase I with DAPI (0.2 µg/ml final concentration). The final volume of the isotype control sample should be 0.5 ml.
NOTE: Ensure that the final density of the primary antibody-stained sample is appropriate for the sorter to be used. A concentration of 5 x 106 cells/ml is routinely used for sorting. - Adjust the volume of the unstained cells to 0.5 ml with PBS/BSA/PS/DNase I. Filter all cells through a 20 µm mesh before sorting and keep the cells on ice in the dark.
- Wash the isotype control and primary antibody-stained samples twice with PBS/BSA/DNase I, as in step 2.2. Resuspend the cells in PBS/BSA/DNase I with DAPI (0.2 µg/ml final concentration). The final volume of the isotype control sample should be 0.5 ml.
- Sorting of Cells.
- Use an 80 µm or larger nozzle for sorting.
NOTE: Murine pancreatic endocrine cells are prone to physical stress. However, murine PCFUs do not appear to be affected by the stress caused by passing through a sorter 7. - Acquire cell events and analysis parameters on the sorter. Gate the cells for forward and side scatter areas to exclude cell debris 7. Gate for forward and side scatter widths to exclude cell doublets. Gate to exclude DAPI+ dead cells 7. Choose gating parameters for the EGFP and CD133-APC signals based on the values from the isotype control cells (Figure 1).
- Collect the sorted cells into 5 ml polystyrene tubes containing 1.5 ml of DMEM/F12 medium supplemented with 5% fetal calf serum (FCS). Centrifuge the sorted populations at 400 x g for 5 min and resuspend in a small volume (~200 µl) of either DMEM/F12 or PBS/BSA/DNase I supplemented with 5% FCS. Keep the cells on ice until use.
- Use an 80 µm or larger nozzle for sorting.
- Count the number of CD133+Sox9-EGFP+ cells obtained from the sorter. Take a 10 µl cell sample and count the cells with a hemacytometer to determine the cell density.
NOTE: Hemacytometer use is recommended because machine counts from the sorter are often unreliable.
3. Plate the Sorted Cells into the Colony Assay Containing Murine ECM Proteins
NOTE: Please refer to the detailed protocols for the plating of cells into the murine ECM-containing colony assay in another JoVE publication 18.
- Prepare Culture Media.
- Prepare a culture medium containing DMEM/F12 medium, 1% (wt/v) methylcellulose, 5% (v/v) murine ECM proteins, 50% (v/v) conditioned medium from mESC-derived pancreatic-like cells, 5% (v/v) FCS, 10 mmol/L nicotinamide, 10 ng/ml human recombinant activin B, 0.1 nmol/L exendin-4, and 1 ng/ml vascular endothelial growth factor-A. Methods for generating the conditioned medium are detailed elsewhere 18.
- Add 750 ng/ml of RSPO-1 to the medium if Dense colony formation is desired.
- Incubate the murine ECM protein culture at 37 °C and 5% CO2 for 3 weeks.
4. Culture the Sorted Cells at 1 Cell Per Well
NOTE: The following procedures apply to manipulating single cells using hand and mouth pipettes. An alternative approach is to purchase a micromanipulator. A typical micromanipulator includes an inverted microscope with a joystick-operated, motorized platform.
- Prepare Glass Pasteur Pipettes.
- Using a Bunsen burner, draw out the tip of a glass Pasteur pipette to a fine point (~30 µm at the opening). The glass Pasteur pipettes should have a cotton stopper to create a barrier to the air flow from the operator.
- Autoclave the flamed glass Pasteur pipettes at 121 °C for 20 min for sterilization.
- Prepare 96-well Plates for Culture.
- Prepare cold semi-solid culture medium according to the previously described protocol 18.
- Dispense the medium into the inner wells of a low-binding flat-bottom 96-well plate at 100 µl/well using a 1 ml syringe. Fill the outer wells with sterile water to maintain humidity.
- Place the culture dish on ice until use.
- Prepare the Sorted Cells.
- Add ~6,000 cells collected from a sorter to 2.5 ml final volume of DMEM/F12/PS containing 10% FCS and 1% methylcellulose in a 5 mL polystyrene tube. Shake vigorously to mix the components. Wait for 5 min or until the bubbles float to the top of the tube. This step does not need to be performed on ice.
- Dispense the cell solution to two 35 mm dishes at 1 ml/dish using a 1 ml syringe with an 18 ½ G needle.
- Spread the cell solution in the dish by gently rocking the dish by hand. Do not allow the semi-solid medium to reach the margin of the 35 mm dish, as the single cells at the margin of the well cannot be observed clearly using an inverted light microscope.
- Prepare the Working Area Around a Microscope.
- Make a solution containing DMEM/F-12 medium and 1% methylcellulose without any cells ('no-cell' medium), and dispense the solution (as described in 4.3.2) into two 35 mm Petri dishes (1 ml per dish).
- Clean and wipe the work area around an inverted phase-contrast microscope with a 70% alcohol solution.
- Assemble the Pipette with the Mouth Piece.
- Pick a sterile glass Pasteur pipette that was prepared in step 4.1. Attach the large end of a 1 ml plastic pipette tip to the top of the glass Pasteur pipette. Attach the small end of the 1 ml plastic pipette tip to one end of a thin-walled rubber tube, and the mouthpiece to the other end of the tube. Use the same glass pipette for picking up multiple cells of the same group.
- Draw up, by mouth suction, a small volume (~10 to 50 µl) of the "no-cell" semi-solid solution made in step 4.4.1 with the glass Pasteur pipette.
NOTE: The semi-solid solution at the narrow opening of the glass Pasteur pipette creates flow resistance and provides a barrier to prevent contamination of the cells to be picked.
- Pick a Single Cell.
- Place the 35 mm dish containing the sorted cells on the microscope stage and remove the lid.
- Find the cells using a 10X objective lens and focus on a suitable cell to be picked.
- Find and place the opening of the pipette tip next to the cell of interest. Apply suction through the mouth.
NOTE: The motion of the cell should be quite slow, so that there is no risk of losing the cell during the process later. The motion of the cell entering the opening of the pipette should be visible. If the flow moves too fast, change to a pipette with a narrower opening.
- Deposit the Single Cell into a Culture Well.
- Once the cell is in the pipette, use the tongue to stop the flow by blocking the opening of the mouth piece. Pause briefly before withdrawing the tip from the semi-solid medium to ensure that the semi-solid medium has stopped flowing.
- Place the tip of the pipette into a well in the 96-well plate prepared in step 4.2. Push the cell out slowly by gently blowing into the mouth piece. Mark the well after the cell is deposited, to avoid plating two cells into a well.
- Ensure that a Single Cell is Placed in a Culture Well.
- Place the pipette tip in the 'no-cell' medium prepared in step 4.4.1. While observing the opening of the tip under the microscope, push out the remaining semi-solid solution. Expect no cell to flow out the tip of the pipette.
- To double check, find the location in the culture well where the single cell was implanted.
- Culture the single cells at 37 °C in 5% CO2 for up to 3 weeks.
5. Pick Individual Colonies from Semi-solid Culture for Microfluidic qRT-PCR Analysis
NOTE: Three weeks after plating sorted CD133+Sox9/EGFP+ cells into the murine ECM-containing colony assay, Ring or Dense colonies are formed 7 (Figure 2). When Ring/Dense colonies are dissociated into single cell suspension and re-plated into laminin hydrogel culture, Endocrine/Acinar colonies are generated after approximately one week 7 (Figure 2). To determine the lineage composition of each colony, microfluidic qRT-PCR analysis is used to detect the expression of lineage markers 7. For pre-amplification, a colony is mixed in a master mix containing a TaqMan probe mix, reaction buffer, and SuperScript III 21. The 48.48 array chip is subsequently used for microfluidic PCR reactions 21.
- Prepare the pre-amplification PCR mix containing 48 probes.
- Pipette 1.5 µl of each TaqMan probe (20x stock) in a 1.5 ml tube. Add 78 µl of TE buffer to adjust the volume to 150 µl.
- Prepare the master mix by following protocols from the manufacturer 21.
- Place 9 µl of master mix into each 0.2 ml thin-walled reaction tube suitable for PCR, and repeat this step for up to 48 tubes.
- Pick single colonies from the semi-solid culture.
NOTE: Ring/Dense colonies are larger than Endocrine/Acinar colonies, with diameters ranging from ~50 to 400 µm 9,11. Therefore, single Ring/Dense colonies are picked using a P10 pipettor outfitted with a 10 µl pipette tip that is bent into a curved shape. For picking the small Endocrine/Acinar colonies (Figure 2), refer to step 4.- Locate a colony of interest under high magnification (e.g., 20X objective lens), reduce the magnification, and aspirate the colony. (Optional) Take a phase-contrast image of the colony at this step to document the visible characteristics of the colony.
- Transfer the colony into the PCR tube containing 9 µl of master mix.
- Pick the next colony. Up to 45 colonies in total may be analyzed per PCR run, leaving 3 spots for controls.
- RNA extraction and cDNA synthesis with pre-amplification.
- Vortex the samples vigorously before performing the thermal reaction.
- Perform the thermal cycling reactions according to the manufacturer's instructions 21. Ring/Dense colonies require 14 cycles, Endocrine/Acinar colonies require 16-20 cycles, and single cells require 22 cycles.
NOTE: Pre-amplification cDNAs can be stored at -20 °C prior to microfluidic PCR analysis.
- Run subsequent PCR reactions, using a microfluidic chip, according to the manufacturer's instructions 21.
6. Whole Mount Immunostaining of Colonies
- Harvest and Fix Colonies.
- Pick up the colonies under the microscope, as in steps 4 or 5.4, and add them to a 4% paraformaldehyde solution. Incubate O/N at 4 °C with gentle shaking.
- Wash the colonies with 1X PBS twice for 10-30 min at RT. Store the fixed colonies at 4 °C in 1.5 ml tubes sealed with paraffin.
- Stain the Colonies with Antibodies.
- Transfer colonies to one well of a 96-well black plate with a clear bottom containing 200 µl of blocking buffer (5% donkey and/or goat serum, 0.1% Triton X-100 in 1x PBS). Incubate O/N at 4 °C with gentle shaking.
- Dilute the primary antibody to a pre-determined concentration (e.g., 1:500 for hamster anti-Mucin 1 antibody) using blocking buffer. Transfer the colonies to a clean well with 200 µl of primary antibody, and incubate O/N at 4 °C with gentle shaking. Wash the colonies 3 times with PBS containing 0.1% Tween 20. Transfer the colonies into a new well with 200 µl of 1x PBS/0.1% Tween 20 for 10 min at RT for each wash.
- Dilute the secondary antibody to a pre-determined concentration (e.g., 1:2,000 for goat anti-Armenian hamster antibody) using blocking buffer. Keep the solution in the dark. Transfer the colonies to clean wells containing 200 µl of secondary antibody, and incubate for 2 hr at RT. Wash the colonies 3 times with PBS/0.1% Tween 20, as in step 6.2.2.
- Counter-stain colonies with DAPI and visualize with confocal microscopy.
- Transfer the colonies into wells with PBS containing 300 nM DAPI and incubate for 5 min at RT.
- Transfer the DAPI-stained colonies to a 35 mm glass-bottom Petri dish for visualization. Place a cover slip on top of the colonies to prevent evaporation.
- Use a confocal microscope to capture the images. To visualize DAPI staining, use two-photon excitation wavelength of 730-950 nm. Use an argon laser with a wavelength of 458 nm to excite fluorochromes with emission wavelengths of 519 and 561 nm. Use a helium-neon laser with a wavelength of 633 nm to excite fluorochromes with emission wavelengths of 665 nm.
7. Dissociate and Re-plate Primary Ring/Dense Colonies into Secondary Colony Assays
NOTE: All procedures should be performed under sterile conditions. Avoid cold shock to the cells in this procedure as much as possible, such as putting cells on ice or washing cells with cold PBS/BSA. Such practices reduce the viability of re-plated cells.
- Prepare Solutions.
- Pre-warm wash buffer (DMEM/F12; P/S; 0.1% BSA) in a 37 °C water bath.
- Pre-warm a 96-well plate (flat bottom; low binding) in a 37 °C incubator. Add 100 µl 100% FCS to one well of the plate and 100 µl 0.25% trypsin-EDTA solution to a second well.
- Collect Colonies.
- Pick and pool a total of 20 or more Ring/Dense colonies in primary cultures using 10 µl pipette tips, as described in step 5.4.
- Place the colonies in warm (at least RT) wash buffer (~1,000 µl) in a 1.5 ml tube and spin at 400 x g for 5 min. Remove the supernatant.
- Dissociate the Colonies into Single-cell Suspensions Using Trypsin.
- Transfer the remaining volume (20 µl or less), which contains the colonies, into the well that contains warm trypsin solution (prepared in 7.1).
- Incubate the plate in a 37 °C incubator (not a water bath) for 1.5 min. Remove the plate and pipette the colonies a few times. Incubate at 37 °C for another 1.5 min.
- Remove the plate and pipette up and down a few more times to break up the colonies. Check under the microscope to see whether the colonies have been dispersed predominantly into single-cell suspension. Avoid over-digestion of the colonies.
- Stop the Trypsin Reaction by Adding FCS.
- Transfer 100 µl of warm FCS into the well that contains the cells. Pipette up and down a few times. Transfer the cells into a 1.5 ml tube containing 1,000 µl warm wash buffer. Centrifuge at 400 x g for 5 min at RT.
- Wash twice with warm buffer.
- Re-suspend the cells in ~200 µl of buffer or culture medium.
- Count the number of cells using a hemacytometer. Keep the cell suspension at RT.
- Re-plate dissociated cells into secondary colony assays containing either murine ECM proteins (5% v/v) or laminin hydrogel (100 µg/ml) and incubate the cells at 37 °C with 5% CO2.
NOTE: For re-plating cells into murine ECM proteins, use 2,500-5,000 cells per well and culture for 2-3 weeks. For re-pating into laminin hydrogel culture, use 10,000-25,000 cells per well and culture for 7-12 days.
Representative Results
Adult pancreatic progenitor cells can be enriched by fluorescence-activated cell sorting (Figure 1). The Sox9/EGFP transgenic mouse line used here was first generated as a result of the GENSAT Brain Atlas Project 19, and the EGFP reporter is under the control of a bacterial artificial chromosome containing ~75 kb upstream and ~150 kb downstream sequences of Sox9 20. In these mice, EGFP labels pancreatic ducts efficiently and specifically 22. Pancreas was procured and dissociated into a single-cell suspension, and stained with anti-CD133 antibodies conjugated with biotin, followed by staining with secondary antibodies conjugated with STV-APC. The resulting cells were analyzed by flow cytometry with appropriate gating parameters (Figure 1). Cells obtained using different gating parameters can be sorted and plated into the methylcellulose-containing colony assays for colony formation. In previous studies, it was noted that colony forming capability is only found in the sorted CD133+Sox9/EGFP+ ductal cells 7.
The colonies formed in the methylcellulose-containing colony assays were classified according to their morphologies, as observed using an inverted, phase-contrast, light microscope (Figure 2). Subsequently, individual colonies were hand-picked and analyzed by microfluidic qRT-PCR analysis for gene expression (Figure 3) or by whole-mount immunostaining for protein expression (Figure 4). In published studies 7,9,11, it was found that many individual colonies expressed lineage markers for duct (mucin-1), acinar (amylase) and endocrine (insulin) cells, indicating that the majority of the initiating PCFUs for these colonies are tri-potent. The proportion of the three cell lineages in each colony, however, was influenced by the types and concentrations of ECM proteins present in the methylcellulose-containing colony assays 7,9. Murine ECM proteins stimulated self-renewal of PCFUs and ductal cell differentiation whereas laminin hydrogel preferentially supported the differentiation of endocrine and acinar cell lineages 7,9,11.
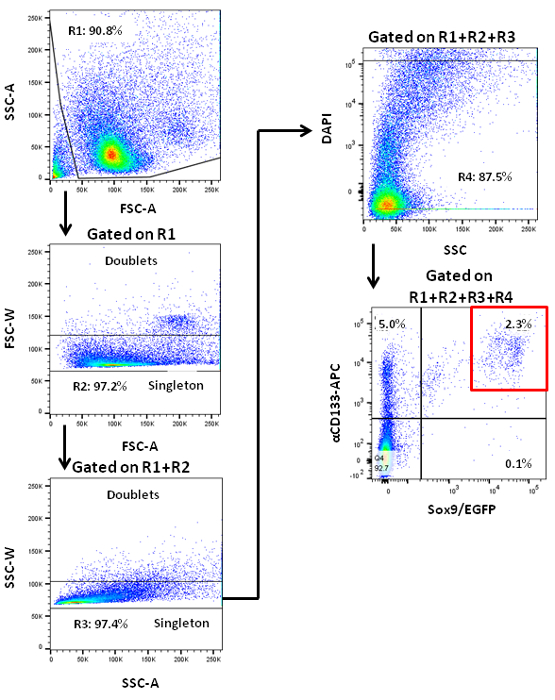
Figure 1. Flow cytometry analysis of dissociated pancreatic cells from adult mice showing staining patterns according to two ductal cell markers, CD133 and Sox9. Sox9/EGFP transgenic mice that contained Sox9 loci-driven EGFP reporter were used. A representative sorting gate (red box) for pancreatic CD133+Sox9/EGFP+ cells is shown. CD133+Sox9/EGFP+ cells are enriched for PCFUs 7. Please click here to view a larger version of this figure.
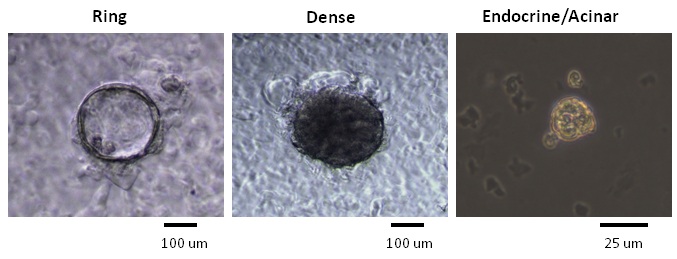
Figure 2. Colonies with different morphologies were generated in methylcellulose-containing colony assays. Representative photomicrographs of Ring, Dense and Endocrine/Acinar colonies from a phase-contrast microscope illuminated by visible light are shown. Ring and Dense colonies are generated when freshly sorted PCFUs are plated into culture medium containing murine ECM proteins and cultured for 3 weeks 7. Endocrine/Acinar colonies are formed after re-plating the dissociated Ring or Dense colony cells into culture medium containing a laminin hydrogel and cultured for ~1 week 7. Scale bars for Ring and Dense colonies = 100 µm. Scale bar for Endo/Acinar colonies = 25 µm. Please click here to view a larger version of this figure.
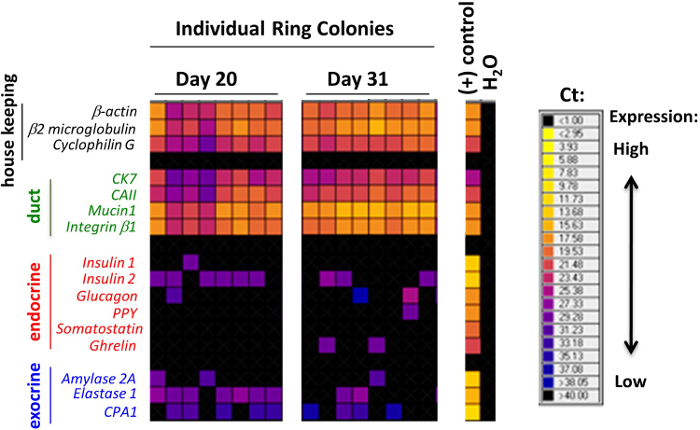
Figure 3. Individual colonies grown in murine ECM proteins expressed markers for ductal, acinar and endocrine cells. Representative results of microfluidic qRT-PCR analysis of individual Ring colonies. Each column represents a single colony. The expression levels of genes are expressed as a heat-map here, with warmer colors indicative of higher expression and colder colors indicative of lower expression. Many of the single colonies express at least one of the genes in each panel for the markers indicative of duct, endocrine or acinar cell lineage. These data demonstrate that many of the individual colonies express tri-lineage markers, and suggest that most of the originating PCFUs are tri-potent. This figure is modified from a previously published figure 7. Please click here to view a larger version of this figure.
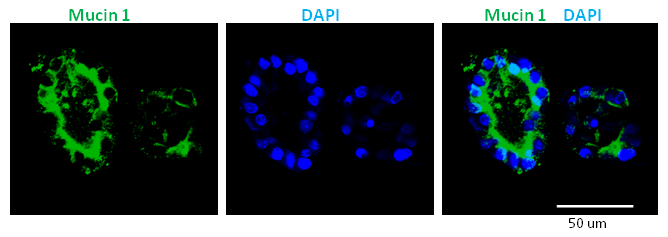
Figure 4. Two Ring colonies expressed a ductal protein marker Mucin 1. Representative results of whole-mount immunostaining of Ring colonies, showing protein expression of a ductal marker Mucin 1 (green color). The nuclei are counter-stained with DAPI (blue color). Many cells in these Ring colonies express Mucin 1 protein. This is consistent with the microfluidic qRT-PCR results showing that Ring colonies grown in murine ECM proteins express high levels (warmer colors in Figure 3) of mucin1 and other ductal cell markers. The scale bar represents 50 µm. Please click here to view a larger version of this figure.
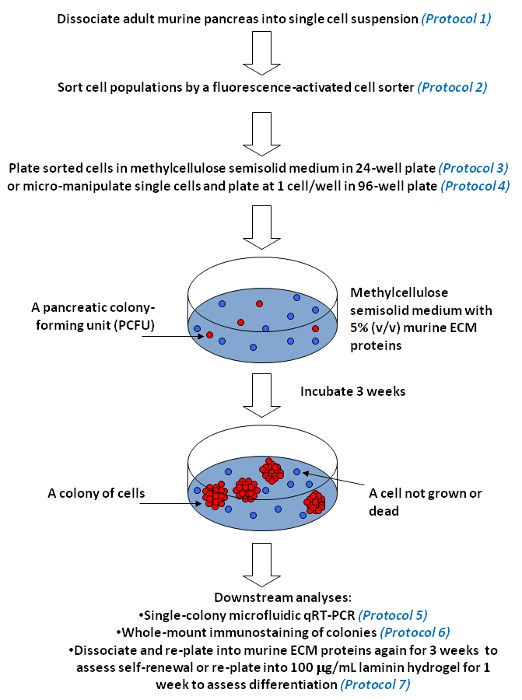
Figure 5. Pancreatic colony assays for measurement of self-renewal and multi-lineage differentiation of individual progenitor cells in culture. Representative workflow is presented. Our pancreatic colony assays contain a viscous material, methylcellulose, so that the culture medium becomes semi-solid. Semi-solid medium restricts the movement and prevents aggregation of single progenitor cells (indicated in red). However, the medium is soft enough to allow a single progenitor cell to self-renew and/or differentiate, and grow into a colony of cells (represented by multiple red circles). In contrast, single cells that do not have colony-forming abilities, i.e., non-progenitor cells (indicated in blue), remain as single cells or die over time in culture. Methylcellulose also allows the inclusion of ECM proteins at low concentrations, such as 5% murine ECM proteins or 100 µg/ml laminin hydrogel. Please click here to view a larger version of this figure.
Discussion
The pancreatic colony assays and single colony analyses described here were inspired by the methylcellulose-containing hematopoietic colony assays that have played major roles in deciphering the biology of hematopoietic progenitor cells in the past decades 23. In these assays (Figure 5), dissociated pancreatic cells are plated into methylcellulose-containing semi-solid media with appropriate growth factors and ECM proteins that support the formation of Ring, Dense or Endocrine/Acinar colonies 7. A single progenitor cell that is capable of giving rise to a colony of cells is termed a PCFU. By characterizing individual colonies using microfluidic qRT-PCR and whole-mount immunostaining, the lineage potentials of the originating PCFUs can be deduced. It was found that a majority of adult murine PCFUs are tri-potent, capable of giving rise to ductal, acinar, and endocrine lineage cells in vitro 7,9. While murine ECM proteins encourage the differentiation of tri-potent PCFU towards duct lineage, the laminin hydrogel allows robust endocrine and acinar cell lineage differentiation 7,9. To assess the in vitro self-renewal capacity of the PCFUs, the Ring or Dense colonies grown in a primary murine ECM colony assay can be dissociated into single-cell suspension and re-plated into a secondary murine ECM colony assay 7. It was found that PCFUs expanded ~500,000 times in murine ECM proteins over 11 weeks 7. The critical steps include the removal of the fat tissues from the dissected pancreata, avoiding over-digestion of pancreatic cells by collagenase, and minimizing cold shock to the dissociated Ring/Dense colony cells before re-plating. Mastering micromanipulation of single cells may take some time; patience and practice are required.
Compared to other pancreatic progenitor cell assays, including 2D culture 24, the suspension "pancreasphere" 25 and the organoid assays 12-15, the major advantages of the methylcellulose-containing colony assays described here are as follows. First, the addition and tuning of ECM proteins to a wide range of concentrations can be easily achieved. This is because methylcellulose is the substance that confers the 3D nature of the semi-solid medium. In contrast, the organoid culture methods depend on the solidification of the murine ECM proteins, which are present at 33% v/v or higher. It is noted that as little as 1% murine ECM protein may inhibit the differentiation of endocrine cells 9, underscoring the importance of ECM protein concentration in the progenitor cell assay. Second, colonies are evenly distributed across the culture well and can be counted accurately. Third, single colonies can be easily handpicked for subsequent analysis. Fourth, the methylcellulose-containing semi-solid medium is easy to maintain, and no medium change is required during the course of the culture. Finally, large numbers of cells (up to 25,000 cells per 24-well plate) can be plated and examined in these colony assays, making them efficient for detecting progenitor cell activities even if they are a minor population among the plated cells.
The pancreatic progenitor cell assays described here are based on functional, but not marker-based, analyses of the pancreatic progenitor cells. This means that pancreatic progenitor cells can be studied without knowing what markers they express. This is important because there is little knowledge about what specific markers the adult pancreatic progenitor cells may express. In addition, markers for embryonic pancreatic progenitor cells may not be adequate for studying the adult progenitors. By using functional analyses, one may begin to identify specific progenitor cell markers by first determining the sub-population that has PCFU activity, such as the CD133+Sox9/EGFP+ cells illustrated here. This can be followed by the identification, using RNA-seq, of genes that are differentially expressed by the PCFU-containing population. The candidate markers may then be tested and verified by assays such as in vitro and in vivo lineage tracing techniques that track the differentiation abilities of the progenitor cells.
The limitations of the in vitro methylcellulose-containing colony assays are threefold. First, although the assays mimic the in vivo microenvironment of the pancreas by providing ECM proteins and growth factors to the pancreatic progenitor cells in 3D space in culture, the conditions are not identical. In addition, the structures of the epithelial cells in vivo are disrupted by dissociation into single cells, which may have important consequences. Therefore, it will be important to verify progenitor cells in follow-up studies using in vivo assays. Second, the culture components contain undefined reagents, such as murine ECM proteins, FCS, and conditioned media 18. These undefined components may affect the function of specific growth factors on the progenitor cells indirectly and more work is needed to create a fully defined set of culture conditions. Finally, lineage tracing experiments demonstrated acinar-to-beta cells 26,27 or alpha cells-to-beta cells 28 conversions in vivo in adult mice. The methylcellulose culture conditions described here may be specific for duct-to-beta cell conversion only. Other unknown culture conditions may be needed for non-duct cells to convert to beta cells in culture.
There are several instances where troubleshooting may be necessary. For example, if dissociated cells are clumped together, use higher doses of DNase I in solutions. DNase I is important to prevent tangling of dissociated pancreatic cells caused by DNA molecules released by the dead cells (see step 1.3.2).
Alternatively, if minced tissues stick in the 10 ml pipette, mince the tissue into smaller pieces. The opening of the 10 ml pipette should be wide enough for the tissue pieces to pass through after collagenase digestion. If pieces get stuck at the tip of the 10 ml pipette, this indicates that the mincing of the tissue by the spring scissors is not sufficient (see step 1.3.4).
Another problem that may arise is no colony growth in murine ECM proteins from dissociated pancreatic cells (i.e., unsorted cells). If this happens, do not attempt to break all of the clusters into single cells during the pancreas dissociation step, as this may result in overdigestion and cell death. If there are large pieces, return the tube to 37 °C for a few (4) min and syringe 7 times or less. Recheck the cells under the microscope. The maximum time for collagenase B treatment is 30 min. Depending on the collagenase used, the optimal time for digestion may differ (see step 1.4.2).
In addition, clumps of murine extracellular matrix proteins in culture wells may be observed after incubation. To avoid this, make sure that all of the culture components that come into contact with murine extracellular matrix proteins are kept cold on ice to avoid premature solidification prior to incubation in 37 °C (see step 3).
In addition, it may be difficult in picking up one cell at a time into a glass Pasteur pipette. A solution to this is to reduce the density of the freshly sorted cells plated in the semi-solid medium to make sure that a cell has enough distance from the other cells. This will avoid picking up multiple cells at the same time. In addition, it is better to place the focus of the microscope near the bottom of the semi-solid medium, as it is easier to pick up single cells by hand near that plane without a micromanipulator (see step 4.6.2).
In addition, it may be unclear whether single cell manipulation is successful. A solution is to practice the single cell micromanipulation before the actual experiment. Once the cell is picked up in the glass Pasteur pipette, transfer the cell to a 35 mm Petri dish containing 1 ml of 1% methylcellulose solution without cells ('no-cell' medium prepared in step 4.4.1). Visualize and place the opening of the tip of the Pasteur pipette in focus under the microscope, and push the cell out slowly. Expect the single cell to slowly appear around the tip of the pipette and then move into the 'no-cell' medium (see step 4.8.2).
Finally, no colony growth may occur in secondary cultures from dissociated primary Ring/Dense colonies. For troubleshooting, ensure that cells from dissociated primary colonies are kept in RT before plating into secondary cultures. For culturing in murine ECM proteins, add the cells last to the tube containing cold culture medium before mixing 18, thereby minimizing the exposure of the cells to cold temperature, which may kill the dissociated cells obtained from primary colonies. However, the re-plating of cells into laminin hydrogel does not need to be performed on ice before 37 °C incubation (see step 7).
In summary, two methylcellulose-containing colony assays have been used to characterize progenitor-like cells from the pancreata of adult 7,11 and postnatal young mice 9,10, as well as from the livers of young mice 10. Future applications will be directed at the identification and characterization of the human colony-forming progenitor cells from cadaveric pancreatic organs.
開示
The authors have nothing to disclose.
Acknowledgements
We thank Lucy Brown and Alexander Spalla from the Analytical Cytometry Core at City of Hope for assistance in sorting. This work is supported in part by National Institutes of Health (NIH) grants R01DK081587 and R01DK099734 to H.T.K., and U01DK089533 to A.D.R., and by National Science Foundation grant NSF-DMR-1206121 and California Institute for Regenerative Medicine grant RB5-07398 to D.A.T. Supports from the Joseph J. Jacobs Institute for Molecular Engineering for Medicine at Caltech to D.A.T., and those from Oxnard Foundation and Ella Fitzgerald Foundation to H.T.K. are also gratefully acknowledged.
Funding: This work is supported in part by National Institutes of Health (NIH) grants R01DK081587 and R01DK099734 to H.T.K., and U01DK089533 to A.D.R., and by National Science Foundation grant NSF-DMR-1206121 and California Institute for Regenerative Medicine grant RB5-07398 to D.A.T. Supports from the Joseph J. Jacobs Institute for Molecular Engineering for Medicine at Caltech to D.A.T., and those from Oxnard Foundation and Ella Fitzgerald Foundation to H.T.K. are also gratefully acknowledged. Research reported in this publication included work performed in the Analytical Cytometry Core and Light Microscopy Digital Imaging Core supported by the National Cancer Institute of the National Institutes of Health under award number P30CA33572.
Study sponsor: The sponsor did not participate in the study design, collection, analysis, or interpretation of data.
Materials
| Murine ECM proteins (Matrigel) | Becton Dickson (Franklin Lakes, NJ, USA) | 354230 | Stock kept at -20oC |
| Laminin Hydrogel | Provided by David Tirrell (Pasadena, CA USA) | Stock kept at -20oC | |
| Methylcellulose | Shinetsu Chemical (Tokyo, Japan) | 1500 centipoise (dynamic viscosity unit equal to 15g/cm/s) (high viscosity) | |
| Dulbecco's Phosphate-Buffered Saline | Mediatech (Manassas, VA, USA) | 21-031-CV | |
| Phosphate Buffered Saline | Gibco (Grand Island, NY, USA) | 15070-063 | |
| 50mL Flacon Conical vial | Corning Inc. (Corning, NY, USA) | 352070 | |
| 100mmx20mm Suspension culture dish | Corning Inc. (Corning, NY, USA) | 430591 | |
| Bovine Serum Albumin | Sigma (St. Louis, MO, USA) | A8412 | |
| Penicillin/Streptomycin | Gibco (Grand Island, NY, USA) | 15070-063 | |
| DNase1 | Calbiochem (Darmstadt, Germany) | 260913 | |
| Collagenase B | Roche (CH-4070, Basel, Schweiz, Switzerland) | 11088831001 | Stock kept at -20oC |
| Anti-mouse CD16/32 | Biolegend (San Diego, CA, USA) | 101310 | low endotoxin, azide free |
| PE-Cy7 Rat IgG2a κ Isotype Control | Biolegend (San Diego, CA, USA) | 400522 | |
| Rat IgG1 κ Isotype Control | eBioscience (San Diego, CA, USA) | 13-4301-82 | |
| Anti-CD133-Biotin | eBioscience (San Diego, CA, USA) | 13-1331-82 | |
| Anti-CD71-PE-Cy7 | Biolegend (San Diego, CA, USA) | 113812 | |
| Streptavidin-Allophycocyanin | Biolegend (San Diego, CA, USA) | 405207 | |
| 4',6-Diamidino-2-phenylindole | Invitrogen (Waltham, MA, USA) | 3571 | Stock kept at -20oC |
| Anti-mucin 1 | Thermo Fisher Scientific (Waltham, MA USA) | HM-1630-P1 | |
| Dylight 649 Goat anti-Armenian Hamster | Jackson Immuno (West Grove, PA, USA) | 127-495-160 | |
| Dulbecco's Modified Eagle Medium: Nutrient Mixture F-12 | Mediatech(Manassas, VA, USA) | 10-092-CV | |
| Fetal Bovine Serum | Tissure Culture Biologicals (Long Beach, CA, USA) | 101 | Stock kept at -20oC |
| Tris Ethylenediaminetetraacetic acid | TEKnova (Hollister, CA, USA) | T0221 | |
| Rneasy Micro Kit | Qiagen (Venlo, Netherlands) | 74004 | |
| QuantiTec Reverse Transcription Kit | Qiagen (Venlo, Netherlands) | 205310 | |
| CellsDirect One-Step qRT-PCR Kit | Ambion/Invitrogen(Grand Island, NY, USA) | 11753-100 | |
| Paraformaldehyde | Santa Cruz Bio (Santa Cruz, CA, USA) | SC-281692 | |
| Goat serum | Jackson Immuno (West Grove, PA, USA) | 005-000-121 | Stock kept at -20oC |
| Donkey serum | Jackson Immuno (West Grove, PA, USA) | 0017-000-121 | Stock kept at -20oC |
| Triton X-100 | Sigma (St. Louis, MO, USA) | T9284 | |
| Trypsin | Sigma (St. Louis, MO, USA) | T-4799 | |
| Ethylenediaminetetraacetic acid | Invitrogen (Waltham, MA, USA) | 15575-020 | |
| Trypsin-EDTA | Life Technologies (Waltham, MA, USA) | 25200-056 | |
| Sterile Water | Gibco (Grand Island, NY, USA) | 15230-147 | Molecular biology grade |
| Pasteur Pipette | Fisher Scientific (Pittsburgh, PA , USA) | 13-678-8B | |
| 40um Filter Mesh | Fisher Scientific (Pittsburgh, PA , USA) | 08-771-1 | |
| 70 um filter mesh | Fisher Scientific (Pittsburgh, PA , USA) | 08-771-2 | |
| TC Plate 96 Well Suspension | Sarstedt | 83.3924 (Previously 83.1835) | |
| 1cc Syringe | Becton Dickson (Franklin Lakes, NJ, USA) | 309659 | |
| 10cc Syringe | Becton Dickson (Franklin Lakes, NJ, USA) | 301604 | |
| 48.48 Dyanmic Array Chip | Fluidigm (San Francisco, CA, USA) | BMK-M-48.48 | |
| Fluidigm GE 48.48 Dynamic Array Sample & Assay Loading Reagent Kit | Fluidigm (San Francisco, CA, USA) | 85000800 | |
| TaqMan Universal PCR Master Mix | Applied Biosystems (Grand Island, NY, USA) | 4304437 | |
| Polyethylene glycol sorbitan monolaurate | Sigma (St. Louis, MO, USA) | P7949 | |
| Glass Bottom Dish | MatTek (Ashland, MA, USA) | P35G-1.5-14-C | 35 mm petri dish with glass bottom |
| Mouth Piece/ Rubber Tubing | Renova Life Inc. (College Park, MD, USA) | MP-SET | |
| Nicotinamide | Sigma (St. Louis, MO, USA) | N0636 | Stock kept at -20oC |
| Vascular Endothelial Growth Factor | R&D Systems (Minneapolis, MN, USA) | 293-VE | Stock kept at -80oC |
| Activin B | R&D Systems (Minneapolis, MN, USA) | 659-AB | Stock kept at -80oC |
| Extendin 4 | Sigma (St. Louis, MO, USA) | E7144 | Stock kept at -20oC |
| Rspondin-1 | R&D Systems (Minneapolis, MN, USA) | 3474-RS | Stock kept at -80oC |
| Falcon 5mL Polystyrene Round-Bottom Tube | Corning Inc. (Corning, NY, USA) | 352054 | |
| PrecisionGlide Needle 18Gx1 1/2 | Becton Dickson (Franklin Lakes, NJ, USA) | 305196 | |
| PrecisionGlide Needle 16Gx1 1/2 | Becton Dickson (Franklin Lakes, NJ, USA) | 305198 | |
| Costar Ultra-Low Attachment Surface 24 well flat bottom plate | Corning Inc. (Corning, NY, USA) | 3473 | |
| Costar 96 Black Well Plate | Corning Inc. (Corning, NY, USA) | 3603 | Flat, clear bottom with lid. Black polystyrene TC-treated microplates |
| Zeiss LSM510 META NLO Axiovert 200M Inverted Microscope | Carl Zeiss AG (Oberkochen, Germany) | ||
| Biomark HD | Fluidigm (San Francisco, CA, USA) | ||
| Aria Special Order Research Product Cell Sorter | Becton Dickson (Franklin Lakes, NJ, USA) |
参考文献
- Gu, G., Brown, J. R., Melton, D. A. Direct lineage tracing reveals the ontogeny of pancreatic cell fates during mouse embryogenesis. Mech Dev. 120, 35-43 (2003).
- Kopp, J. L., et al. Sox9+ ductal cells are multipotent progenitors throughout development but do not produce new endocrine cells in the normal or injured adult pancreas. Development. 138, 653-665 (2011).
- DiGiusto, D. L., et al. RNA-based gene therapy for HIV with lentiviral vector-modified CD34(+) cells in patients undergoing transplantation for AIDS-related lymphoma. Science translational medicine. 2, 36-43 (2010).
- Inada, A., et al. Carbonic anhydrase II-positive pancreatic cells are progenitors for both endocrine and exocrine pancreas after birth. Proc Natl Acad Sci U S A. 105, 19915-19919 (2008).
- Solar, M., et al. Pancreatic exocrine duct cells give rise to insulin-producing beta cells during embryogenesis but not after birth. Dev Cell. 17, 849-860 (2009).
- Ku, H. T. Minireview: pancreatic progenitor cells–recent studies. Endocrinology. 149, 4312-4316 (2008).
- Jin, L., et al. Colony-forming cells in the adult mouse pancreas are expandable in Matrigel and form endocrine/acinar colonies in laminin hydrogel. Proc Natl Acad Sci U S A. 110, 3907-3912 (2013).
- Fu, X., et al. MicroRNA-26a targets ten eleven translocation enzymes and is regulated during pancreatic cell differentiation. Proc Natl Acad Sci U S A. 110, 17892-17897 (2013).
- Ghazalli, N., et al. Postnatal Pancreas of Mice Contains Tripotent Progenitors Capable of Giving Rise to Duct, Acinar, and Endocrine Cells In Vitro. Stem Cells Dev. , (2015).
- Jin, L., et al. Colony-forming progenitor cells in the postnatal mouse liver and pancreas give rise to morphologically distinct insulin-expressing colonies in 3D cultures. Rev Diabet Stud. 11, 35-50 (2014).
- Jin, L., et al. In Vitro Multilineage Differentiation and Self-Renewal of Single Pancreatic Colony-Forming Cells from Adult C57Bl/6 Mice. Stem Cells Dev. , (2014).
- Huch, M., et al. Unlimited in vitro expansion of adult bi-potent pancreas progenitors through the Lgr5/R-spondin axis. Embo J. 32, 2708-2721 (2013).
- Greggio, C., et al. Artificial three-dimensional niches deconstruct pancreas development in vitro. Development. 140, 4452-4462 (2013).
- Lee, J., et al. Expansion and conversion of human pancreatic ductal cells into insulin-secreting endocrine cells. Elife. 2, e00940 (2013).
- Dorrell, C., et al. The organoid-initiating cells in mouse pancreas and liver are phenotypically and functionally similar. Stem Cell Res. 13, 275-283 (2014).
- Ku, H., Yonemura, Y., Kaushansky, K., Ogawa, M. Thrombopoietin, the ligand for the Mpl receptor, synergizes with steel factor and other early acting cytokines in supporting proliferation of primitive hematopoietic progenitors of mice. Blood. 87, 4544-4551 (1996).
- Ku, H. T., et al. Insulin-expressing colonies developed from murine embryonic stem cell-derived progenitors. Diabetes. 56, 921-929 (2007).
- Winkler, M., et al. A quantitative assay for insulin-expressing colony-forming progenitors. J Vis Exp. , e3148 (2011).
- Gong, S., et al. A gene expression atlas of the central nervous system based on bacterial artificial chromosomes. Nature. 425, 917-925 (2003).
- Formeister, E. J., et al. Distinct SOX9 levels differentially mark stem/progenitor populations and enteroendocrine cells of the small intestine epithelium. Am J Physiol Gastrointest Liver Physiol. 296, G1108-G1118 (2009).
- Seymour, P. A., et al. A dosage-dependent requirement for Sox9 in pancreatic endocrine cell formation. Dev Biol. 323, 19-30 (2008).
- Ogawa, M. Differentiation and proliferation of hematopoietic stem cells. Blood. 81, 2844-2853 (1993).
- Suzuki, A., Nakauchi, H., Taniguchi, H. Prospective isolation of multipotent pancreatic progenitors using flow-cytometric cell sorting. Diabetes. 53, 2143-2152 (2004).
- Seaberg, R. M., et al. Clonal identification of multipotent precursors from adult mouse pancreas that generate neural and pancreatic lineages. Nat Biotechnol. 22, 1115-1124 (2004).
- Zhou, Q., Brown, J., Kanarek, A., Rajagopal, J., Melton, D. A. In vivo reprogramming of adult pancreatic exocrine cells to beta-cells. Nature. 455, 627-632 (2008).
- Baeyens, L., et al. Transient cytokine treatment induces acinar cell reprogramming and regenerates functional beta cell mass in diabetic mice. Nat Biotechnol. 32, 76-83 (2014).
- Thorel, F., et al. Conversion of adult pancreatic alpha-cells to beta-cells after extreme beta-cell loss. Nature. 464, 1149-1154 (2010).
