The protocol we describe here represents an optimized P. syringae syringe infiltration assay to quantitatively evaluate the immune response in Arabidopsis plants. As illustrated in Figure 1, the syringe infiltration of Psm ES4326 is followed by pathogen extraction and quantification via serial dilutions and colonies enumeration.
As described in Step 3 within the protocol text, Enhanced Disease Susceptibility (EDS) against Psm ES4326 can be assessed by infection with an inoculum with OD=0.0002. As shown in Figure 3, the highly susceptible npr1-1 mutants have approximately 2.5 log (300 times) more pathogen growth compared to the wild-type Col-0 plants. Depending on the experimental conditions, the npr1-1 plants may support up to 3.0 log more bacterial growth than Col-0, with most common results within the range of 1.5-2.5 log. Therefore, this EDS assay provides a broad window of difference, in which the researchers may be able to place their Arabidopsis mutants and transgenics to identify candidate genes potentially involved in the plant immune response.
In addition to determining EDS, Psm ES4326 syringe infiltration assay can be modified to dissect different layers of immune response. To evaluate Salicylic Acid-Triggered Immunity, external application of the chemical Sodium Salicylate is used to trigger the immune response, which is quantified by the pathogen growth (Figure 4A). The loss of NPR1, which functions as the SA receptor and major transcriptional co-regulator of SA-dependent target genes, leads to insensitivity to exogenous application of salicylic acid derivative. This insensitivity to SA is demonstrated by the unaltered pathogen growth in the npr1-1 Sodium Salicylate pre-treated plants in contrast to a marked reduction in Col-0 plants (20 times less pathogen upon Sodium Salicylate application) (Figure 4B).
To characterize the MAMP-Triggered Immunity, pre-treatment with flg22 or elf18 was performed as demonstrated in Figure 5A. To characterize flagellin-triggered immunity, Col-0 and fls2 mutant plants are used. FLS2, a membrane-localized receptor-like kinase, recognizes flg22 and triggers MTI9. As demonstrated in Figure 5B, the flg22-treated Col-0 plants supported a ~ 1 log (10 times) reduction in the bacterial population, while the fls2 mutant plants failed to trigger the bacterial growth restriction effect. Similarly, the loss of EF-Tu receptor EFR in the efr mutant plants leads to insensitivity to elf18 pre-treatment as demonstrated by the unaltered pathogen growth following the elf18 pre-treatment (Figure 5B).

Figure 1. Schematic representation of Psm ES4326 syringe infiltration assay on Arabidopsis plants. Leaves number 5 and 6 of adult soil-grown Arabidopsis plants are marked and infected with Psm ES4326 through syringe infiltration. After the emergence of disease symptoms, detach leaves and harvest leaf discs for tissue homogenization. Well-homogenized tissue is serially diluted in 96-well plates before transferring onto a bacteria counting plate. After colonies emergence on the plate, count the number of bacteria and process the data to generate a graph. Please click here to view a larger version of this figure.
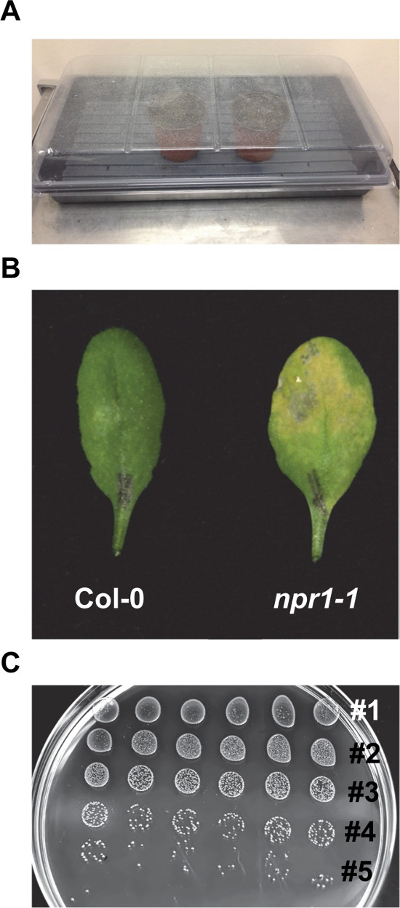
Figure 2. Representative procedures of the EDS assay. (A) Pots are covered with a water-sprayed transparent dome after sowing seeds. (B) Representative symptoms on Col-0 and npr1-1 leaves subjected to the EDS assay (OD600nm = 0.0002) 3 days post inoculation. Note severe chlorosis on npr1-1 and the nearly normal appearance of Col-0. (C) Serial dilutions of Psm ES4326 growing on KB (50 µg/ml streptomycin) media plate after ~ 45 hR of incubation. Five dilutions are visible. Please click here to view a larger version of this figure.
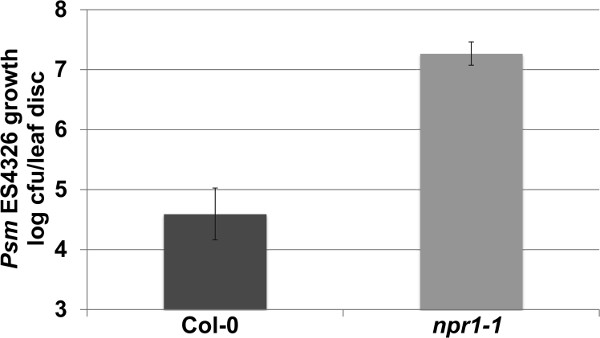
Figure 3. Representative results of the EDS assay. Psm ES4326 growth (colony forming units – cfu/leaf disc, expressed on a log scale) was quantified in 4-week-old Col-0 and npr1-1 plants 3 days post inoculation (OD600nm = 0.0002). Error bars represent 95% confidence intervals of the mean (n = 6). Please click here to view a larger version of this figure.
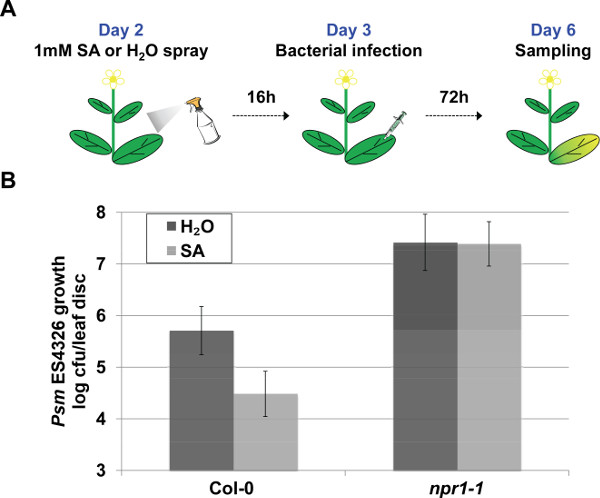
Figure 4. Representative procedure and results of the STI assay. (A) Schematic representation of the Salicylic Acid-Triggered Immunity (STI) assay. (B) Salicylic Acid-Triggered Immunity was quantified based on pathogen growth in plants pre-treated with 1mM Sodium Salicylate or H2O 16 hr prior to Psm ES4326 syringe infiltration (OD600nm = 0.001). Pathogen growth was quantified 3 days post inoculation. Error bars represent 95% confidence intervals of the mean (n = 6). Please click here to view a larger version of this figure.
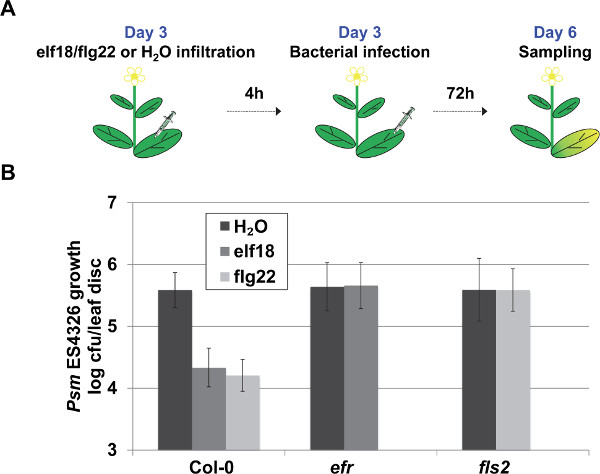
Figure 5. Representative procedure and results of the MTI assay. (A) Schematic representation of the MAMP-Triggered Immunity (MTI) assay. (B) MTI was quantified by pathogen growth in plants pre-treated with 1µM solution of flg22, elf18 or H2O (as control) 4 h prior to Psm ES4326 syringe infiltration (OD600 nm = 0.001). Pathogen growth was quantified 3 days post inoculation. Error bars represent 95% confidence intervals of the mean (n = 6). Please click here to view a larger version of this figure.
| Issue | Possible Causes | Recommended Actions |
| No bacteria growth in the liquid medium after O/N culture | Reduced bacteria activity | Initiate liquid culture from a newly streaked plate |
| Circular syringe impression presents on the leaf after syringe infiltration | Too much pressure during infiltration | Reduce the pressue during infiltration |
| Partial syringe impression presents on the leaf after syringe infiltration | Inappropriate positioning of the syringe | Adjust the syringe to be positioned vertical against the leaf surface |
| Leaf wilts wthtin few hours after infiltration | Too much pathogen solution is infiltrated into the leaf | Stop infiltrating immediately after the entire leaf surface turns darker green in color |
| No disease symptoms three days after infection | Humidity is too low for pathogen to proliferate | Increase the humidity where infected plants are maintained |
| Pathogen enters necrotrophic stage at or before 3 dpi | Incorrect concentration of pathogen solution | Use OD600nm = 0.0002 for EDS assay; confirm dilution using indepedent spectrophotometer |
| Droplets merge on the top of bacteria counting plate | Bacteria counting plate is not appropriately dried | Pre-dry the plate O/N before use |
| Pathogen dilution does not represent 10-fold reduction | Dilution is not accurately performed | Use a well calibrated multi-channel pipette for transferring liquid; confirm that all liquid is dispensed |
| Pathogen growth in the wild type exceeds 8 log cfu/leaf disc | Pathogen overgrew within the infected tissue – sampling performed too late | Sample infected tissue after the emergence of chlorosis |
| Big variation among technical replicates | Plants are not at the same developmental stage or infiltrated leaf number is inconsistent | Only use plants within the same developmental stage for infection. Confirm synchronous germination. Infect consistent leaf number among different plants |
Table 1. Troubleshooting of the syringe infiltration assay.
Please click here to view Table 2.
Table 2. Spreadsheet for statistical analysis of pathogen growth data.
