In this first example, we describe the approach for quantifying initial levels of DNA damage in human lymphoblastoid cells after exposure to oxidative damage using H2O2. In order to assess H2O2-induced base lesions and single strand breaks, alkaline comet conditions are used. After loading is complete and cells have been encapsulated with the agarose overlay, a bottomless 96-well plate is pressed onto the surface to create 96-compartments on the chip. Each of the 96-wells can be dosed with a different type or concentration of chemicals. Here four different doses of H2O2 were used and 1x PBS was used as the negative control. Representative images of arrayed comets are shown in Figure 1A, where non-treated (top), 50 μM (middle) and 100 μM (bottom) H2O2-exposed samples demonstrated different levels of comet tails. Analysis of comet images can be performed using commercially available software, which generally quantifies damage levels based on the total fluorescence intensity and migration distance of each comet tail. Measurements are commonly performed on 50 to 100 comets per condition and the median value (either % tail DNA or tail length) is calculated. Figure 1B illustrates the percentage tail DNA response curve analyzed from the TK6 lymphoblastoid cell comets exposed to four different concentrations of H2O2. The consistency among independent experiments demonstrates the efficacy of this approach in assessing DNA damage response from various levels of chemical treatments in cells.
To learn about variation among samples (e.g., among macrowells) and among repeats of the same experiment, inter-sample and inter-experimental variability was plotted in Figure 2A and 2B respectively. Within each experiment, each well of the 96-well chip is equivalent to a different sample and hence the well-to-well variation reveals the inter-sample variation of the device. Here, TK6 cells loaded in 12 wells of the 96-well chip were treated with the same dose of H2O2 and immediately lysed after treatment. All other experimental steps were performed in parallel and the medians of at least 50 comets from each macrowell were plotted in Figure 2A. The average of the medians is 77.53% DNA in tail, with standard deviation is 3.99%. From these data, we compute that the coefficient of variation among samples is 5.2%, which is several fold lower than the traditional assay, demonstrating the improved robustness of this assay6,16. To learn about the reproducibility of the assay, we exposed TK6 cells in a chip with 75 μM H2O2 and analyze the immediate level of DNA damage. This experiment was repeated six times and data of each experiment was plotted in Figure 2B. Under identical experimental conditions, we obtained results ranging from 41.76% to 57.87%, shown as grey bars in the figure. The average of six repeats is 49.57%, with standard error of the mean of only 2.04%. The low variation among repeats implies that the comet assay performed on this novel device is highly reproducible.
To evaluate repair kinetics, cells are allowed to repair their DNA in fresh media after treatment. Lysing macrowells at various time points reveals the change in DNA damage over time. An example is shown in Figure 3. In this experiment, TK6 cells were first encapsulated in the chip, treated with 50 μM H2O2, and allowed to repair up to 60 min post-exposure. Cells were lysed at 0, 20, 45 and 60 min respectively. Representative comet images at different time points are shown in Figure 3A, and the change in DNA damage levels over time is plotted in Figure 3B. This data revealed that nearly all of the damage is repaired within 45 min post H2O2 treatment. Many variables can affect the rate of repair, including the type of cell line and chemical agent used. Hence researchers can make alterations to the protocol (i.e., extending repair time) to accommodate additional needs in their investigations. Here we provide another example of repair experiment using this chip, wherein TK6 cells are challenged with a different genotoxic agent N-Methyl-N’-Nitro-N-Nitrosoguanidine (MNNG) is an alkylating agent known to induce base lesions including 7-methylguanine and 3-methyladenine. These lesions are converted to abasic sites either by spontaneous depurination or by enzymatic removal by DNA glycosylases. The resulting abasic site can be cleaved under alkaline conditions and thus detected on the chip. Initial exposure causes a significant increase in damage, evident from the long tails, and over time these tails are reduced as the cells restore intact DNA during repair. Although alkylation and oxidative damage are both primarily repaired by the base excision repair pathway, the amount of lesions generated and the proteins involved in repair vary. These variations can lead to different rates of conversion to abasic sites, as well as different rates of repair of the resulting abasic sites. In Figure 4, repair kinetics of TK6 cells exposed to 0.1, 1, and 3 µg/ml of MNNG are shown. In this experiment, we extended the experiment time to 2 hr post-exposure because MNNG-induced damage appears to persist longer and is cleared at a slower rate compared to H2O2-induced damage.
Analysis of DSBs using neutral conditions is very similar to the protocol for the analysis of single-strand lesions using the alkaline conditions. To show initial damage levels, TK6 cells were exposed to variable levels of gIR, and the resulting cells were lysed and electrophoresed at a neutral pH (Figure 5A). It is noteworthy that the morphology of the comet tails is different from the alkaline assay conditions. To achieve observable fragmented DNA using this assay, the IR dose used is significantly higher (0-100 Gy) than the dose required to see damage using the alkaline conditions. In addition, we found that the tail length is the most sensitive parameter in reflecting the extent of DNA damage when using the neutral comet assay7. The analyzed dose response curve is illustrated in Figure 5B. Repair of DNA double strand breaks in cells can also be assessed, and has been shown by Weingeist et. al.7. Taken together, the neutral CometChip offers a valuable tool for high throughput analysis of DNA DSBs.
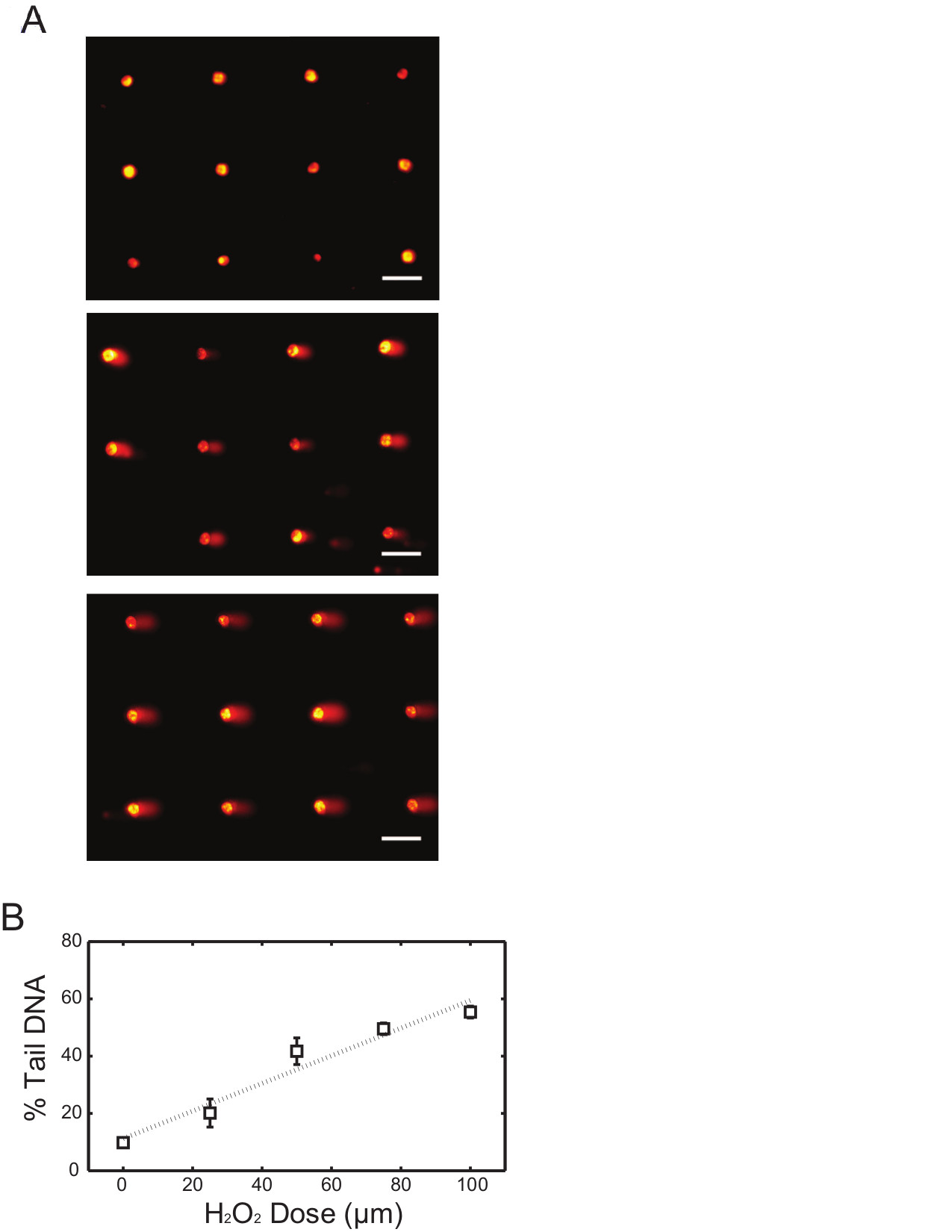
Figure 1. H2O2 dose response of TK6 cells using alkaline comet assay. (A) Arrayed microwell comets from untreated TK6 lymphoblasts (top) and TK6 cells exposed to 50 μM (middle) and 100 μM (bottom) H2O2 for 20 min at 4 °C. Scale bar is 100 μm. (B) H2O2 dose response of TK6 cells exposed to various level of H2O2. Each data point is the average of three independent experiments, where the median percentage tail DNA of at least 50 individual comets was obtained in each experiment. Error bars represent the standard error of the mean from three independent experiments. Please click here to view a larger version of this figure.
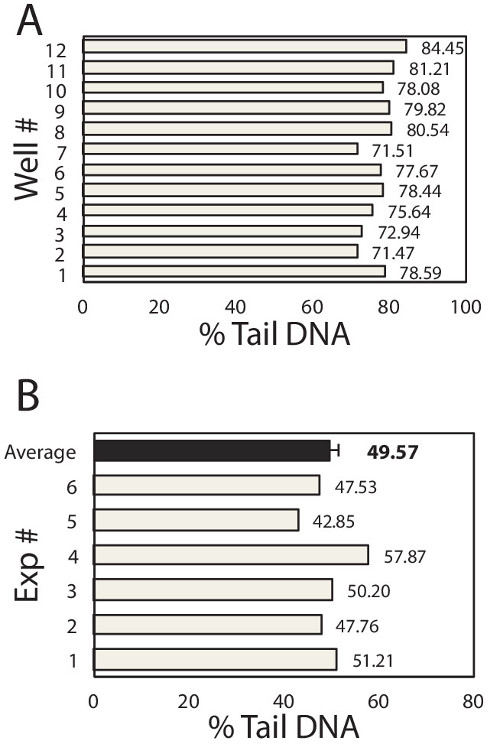
Figure 2. Inter-Sample and Inter-Experimental Variability. (A) Sample-to-Sample variation. TK6 human lymphoblast cells were loaded into 12 wells of the 96-well chip. Each well was exposed to 100 μl of 50 μM H2O2 for 20 min at 4 °C. Cell were immediately lysed and assayed for % Tail DNA. Data of each well is shown here. Each box represents the median at least 50 individual comets from each well. The mean of 12 wells is 77.53%, standard deviation is 3.99%, and the coefficient of variation is computed to be 5.2%. (B) Experiment-to-Experiment variation. TK6 human lymphoblast cells were loaded into the 96-well chip, exposed to 100 μl of 75 μM H2O2 for 20 min at 4 °C. Cell were immediately lysed and assayed for % Tail DNA. The same experiment was repeated 6 times and data of each repeat is shown as a grey bar. Each box represents the median % tail DNA of at least 100 individual comets from each repeat. The average of 6 repeats is 49.57%, shown as the back bar here. The standard deviation of 6 repeats is 4.99%, and the standard error of the mean is computed to be 2.04%, represented as the error bar in the figure.
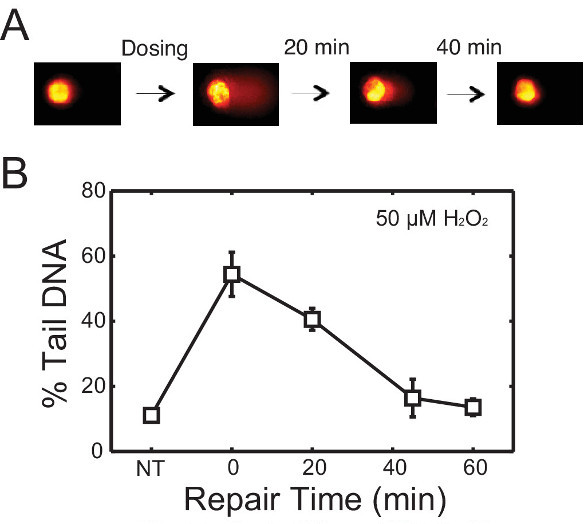
Figure 3. Repair of H2O2-induced damage in TK6 lymphoblasts. (A) Schematic of repair study with representative comets. TK6 cells are treated with damaging agents and allowed to repair in media at 37 °C before lysis. (B) Repair kinetics of TK6 cells after treatment with 50 μM H2O2 for 20 min at 4 °C. Each data point is the average of three independent experiments, where the median percentage tail DNA of at least 50 individual comets was obtained in each experiment. Error bars represent the standard error of the mean from repeating experiments. Please click here to view a larger version of this figure.
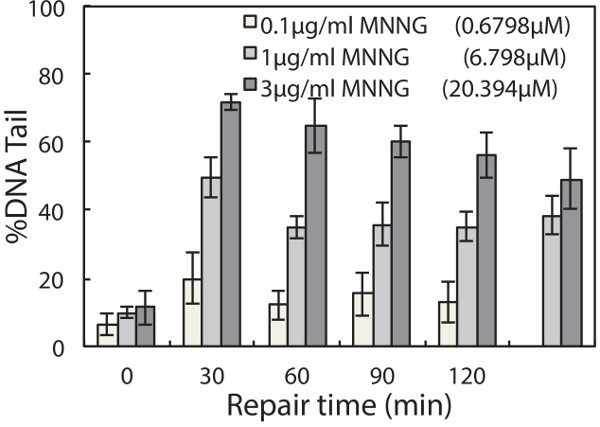
Figure 4. Repair of MNNG-induced damage in TK6 lymphoblasts. TK6 cells are treated with 0.1, 1, and 3 µg/ml MNNG for 30 min at 4 °C and allowed to repair in media at 37 °C up to 120 min post exposure. The median percentage tail DNA of at least 50 individual comets was obtained for each of three independent experiments. Error bars represent the standard error of the mean from three independent experiments.

Figure 5. IR dose response of TK6 cells using neutral comet assay. (A) Arrayed microwell comets from untreated TK6 lymphoblasts (top) and TK6 cells exposed to 40 Gy (middle) and 80 Gy (bottom) gamma irradiation. Scale bar is 100 μm. (B) IR dose response of TK6 cells exposed to various level of gamma irradiation. Each data point represents the median tail length (μm) of at least 300 comets pooled from six macrowells on the chip. Please click here to view a larger version of this figure.
