1. Genotyping of Arabidopsis Plant Individuals with Direct Protocol
- To begin the dCAPS genotyping assay directly on the Arabidopsis plant leaf, first formulate 20 or 50 μl PCR reactions using the Phire Plant Direct PCR Kit as described in Table 1.
- Next, cut a 0.50 mm punch from an Arabidopsis plant leaf using the Harris Uni-Core and Harris Cutting Mat. Holding the puncher firmly, press the cutting edge into the tissue and rotate the puncher back and forth.
- Press the plunger to eject the punch disc into a PCR reaction mixture. Ensure that the sample drops into the PCR solution and does not stick to the tube walls.
- Clean the cutting edge of the puncher between each sample to prevent cross-contamination by dipping it into 2% NaClO solution. Press the plunger up and down a few times and wipe the cutting edge with a clean paper towel. The cutting mat should also be rinsed between samples.
- Next, employ a Thermo Scientific Piko 24-well Thermal Cycler and Thermo Scientific Piko PCR Plate to perform the PCR reactions using the cycling conditions described in Table 2. The PCR reactions can also be performed in conventional PCR thermal cyclers.
- After PCR, spin down the plant material. Transfer 5 μl of the supernatant to a new microcentrifuge tube. Add 4 μl of water and 1 μl of restriction enzyme, SspI. Mix gently and spin down briefly to collect contents in the bottom of the tube. Incubate the reaction mixture for one hour at 37 °C. Inactivate the restriction enzyme by incubating at 65 °C for 20 min.
- When amplifying DNA directly from plant tissues, the PCR products include plant and PCR-derived components that may interfere with the restriction digestion enzyme. Therefore, it may be necessary to either dilute (e.g. 1:2 or 1:3 in water) or purify the PCR product before the subsequent digestion, for example by using a suitable commercial kit such as Thermo Scientific GeneJET PCR Purification Kit.
- Following restriction enzyme digestion, analyze the resulting fragments on an agarose gel. Add 2.5 μl of 5x loading buffer to the reaction and perform agarose gel electrophoresis with 10 μl of the resulting mixture.
2. Amplifying Specific DNA Fragments from 15-year-old Oak Leaves using the Dilution Protocol
- To begin, cut a 2 mm punch of oak leaf.
- Place the sample into 20 μl of Dilution Buffer supplied with the Phire Plant Direct PCR Kit. Clean the cutting edge of the puncher and cutting mat as before.
- Crush the leaf sample with a 100 μl pipette tip by pressing it briefly against the tube wall. Depending on the plant material, the dilution protocol sometimes works better without crushing the sample.
- Spin the samples briefly in a microcentrifuge until the reactions are in the bottom of tubes.
- The supernatant can be used directly in the PCR, or it can be diluted 1:10 or 1:100 in sterile water depending on the sample type. Use 0.5 μl to 1 μl of the supernatant or dilution as a template for a 20 μl PCR reaction. Prepare the reaction as described in Table 1.
- Once the reactions are prepared, employ a Thermo Scientific Piko 24-well Thermal Cycler and Thermo Scientific Piko PCR Plate to perform the PCR reactions using the cycling conditions described in Table 2. The PCR reactions can also be performed in conventional PCR thermal cyclers.
- After PCR, add 5 μl of 5x loading buffer to the reaction and perform agarose gel electrophoresis with 15 μl of the resulting mixture.
3. Genotyping of Transgenic Mice using the Dilution Protocol
- Begin the dilution protocol on transgenic mice by placing a 2 mm punch of the mouse ear in 20 μl of Dilution Buffer containing 0.5 μl of DNARelease Additive supplied with the Phire Animal Tissue Direct PCR Kit.
- Incubate the samples at room temperature for 2 min, followed by 2 min incubation at 98 °C.
- Spin the samples briefly in a microcentrifuge until the reactions are in the bottom of tubes.
- Use 1 μl of supernatant as the template for a 20 μl PCR reaction, prepared as described in Table 3. Any supernatant that is not to be used right away can be transferred to a new tube and stored at -20 °C.
- Next, employ a Thermo Scientific Piko 24-well Thermal Cycler and Thermo Scientific Piko PCR Plate to perform the PCR reactions using the cycling conditions described in Table 4. Again, the PCR reactions can also be performed in conventional PCR cyclers.
- Following PCR, add 5 μl of 5x loading buffer to the reaction and perform agarose gel electrophoresis with 15 μl of the resulting mixture.
4. Representative Results
In the dCAPS technique the restriction site within the SNP of interest is either introduced or destroyed using a PCR primer with one or more mismatches to the target DNA. Genotyping of Arabidopsis plant individuals was performed with the dCAPS technique directly from leaf punches. The amplified products were digested and the resulting fragments revealing the genotype of each individual were analyzed by gel electrophoresis (Figure 2).
In this example, the 160 bp PCR products containing the SNP site of interest (G or A allele) in the Arabidopsis genome were amplified from plant leaf punches with the Phire Plant Direct PCR Kit. The forward primer contained one mismatch at the 3′ end, creating an SspI-specific restriction site in the target DNA, which included the SNP of interest (A allele) but not in the other allele of the SNP (Figure 2B).
Dilution protocols are necessary for challenging plant material, such as the oak leaf, due to its high concentration of phenolic compounds. Using the dilution protocol described here, the 297 bp fragment of chloroplastic DNA was amplified using the 3-step protocol. Oak leaf specimens with different dilutions are shown in Figure 3, in addition to positive and negative controls.
The Phire Animal Tissue Direct PCR Kit was employed with a challenging protocol where only one primer set was used for amplification of two fragments with a large size difference, resulting in abundant yields of both correctly sized products of 1,500 bp for transgenic mice and 200 bp for wild type mice (Figure 4). The weaker upper band with heterozygote mice is due to the competition of both templates (alleles) for the same primer pair; however, the genotyping results are unambiguous.
The stability of the samples prepared using the dilution protocol was also tested. The results show that the dilution samples can be stored at -20 °C for at least one year. Furthermore, repeated freeze/thaw cycles did not significantly affect the reaction (Figure 5). Also shown is the robust amplification from mouse ear tissue samples using Phire DNA Polymerase and the Direct PCR method compared to hot start Taq polymerase and purified DNA (Figure 6).
| Components | 20 μl reaction | 50 μl reaction | Final Conc. |
| H2O | add to 20 μl | add to 50 μl | |
| 2x Phire Plant PCR Buffer | 10 μl | 25 μl | 1x |
| primer A | X μl | X μl | 0.5 μM |
| primer B | X μl | X μl | 0.5 μM |
| Phire Hot Start II DNA Polymerase | 0.4 μl | 1 μl | |
| Sample/direct protocol | 0.50 mm punch | ||
| Sample/dilution protocol | 0.5-1 μl |
Table 1. Reaction conditions for Plant PCR used in this protocol.
| Cycle Step | Temp. | Time | Cycles |
| Initial denaturation | 98 °C | 5 min | 1 |
| Denaturing Annealing* Extension |
98 °C X °C 72 °C |
5 sec 5 sec 20 sec |
40 |
| Final Extension | 72 °C 4 °C |
1 min hold |
1 |
Table 2. Cycling conditions for Plant PCR used in this protocol.
* The Tm calculator at www.thermoscientific.com/pcrwebtools is recommended when determining the Tm for primers to be used with Phire Hot Start II DNA Polymerase. The recommended annealing temperature for primers ≤ 20 nt is equal to the lower Tm given by the webtool. For primers > 20 nt, use an annealing temperature 3 °C higher than the lower Tm given by the webtool.
| Components | 20 μl reaction | Final Conc. |
| H2O | add to 20 μl | |
| 2x Phire Animal Tissue PCR Buffer | 10 μl | 1x |
| primer A | X μl | 0.5 μM |
| primer B | X μl | 0.5 μM |
| Phire Hot Start II DNA Polymerase | 0.4 μl | |
| Sample/dilution protocol | 1 μl |
Table 3. Reaction conditions for Animal Tissue PCR used in this protocol.
| Cycle Step | Temp. | Time | Cycles |
| Initial denaturation | 98 °C | 5 min | 1 |
| Denaturing Annealing* Extension |
98 °C X °C 72 °C |
5 sec 5 sec 20 sec ≤ 1 kb 20 sec/kb > 1 kb |
40 |
| Final Extension | 72 °C 4 °C |
1 min Hold |
1 |
Table 4. Cycling conditions for Animal Tissue PCR used in this protocol.
* The Tm calculator at www.thermoscientific.com/pcrwebtools is recommended when determining the Tm for primers to be used with Phire Hot Start II DNA Polymerase. The recommended annealing temperature for primers ≤ 20 nt is equal to the lower Tm given by the webtool. For primers > 20 nt, use an annealing temperature 3 °C higher than the lower Tm given by the webtool.
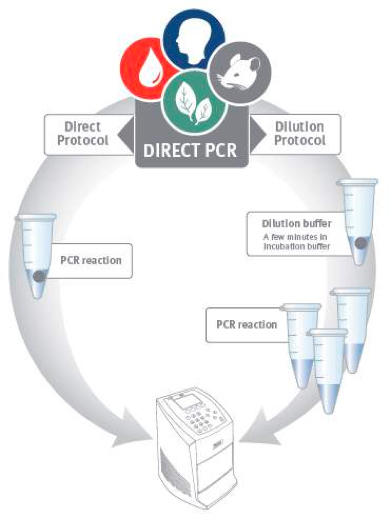
Figure 1. Direct PCR workflow. Direct PCR can be performed using two alternative protocols. In the direct protocol a tiny amount of sample is added directly to the PCR reaction. The dilution protocol employs a brief pre-incubation step before PCR to release DNA from the sample material.
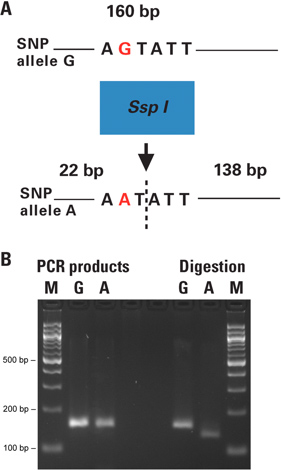
Figure 2. Genotyping of Arabidopsis plant individuals with dCAPS technique directly from leaf punches. A) Figure 2A shows the principle of the dCAPS assay performed. The restriction site within the SNP of interest is either introduced or removed by PCR using a primer with one or more mismatches to the target DNA. The PCR products are digested and the resulting fragments reveal the genotype of each individual when analyzed by electrophoresis. B) 0.50 mm punches from plant leaves were placed directly in 50 μl PCR reactions. The 160 bp PCR products containing the SNP site (G or A allele) were amplified with primers introducing a unique SspI site to the A-allele. The unpurified PCR products were digested with SspI restriction enzyme. Resulting fragments were analyzed on a 3% agarose gel. Lane M is the size marker; lanes A and G correspond to the SNP alleles of each sample. The obtained results were confirmed by conventional analysis of DNA extraction followed by PCR and restriction digestion (data not shown). The negative control reactions without template DNA were included in test set up and run on a separate agarose gel in parallel with the actual PCR reactions before performing the digestions (data not shown).
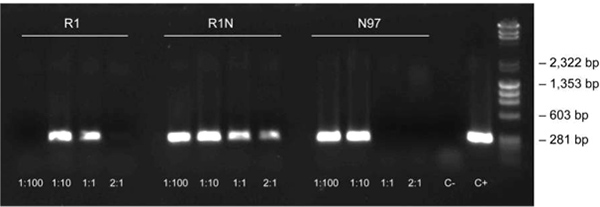
Figure 3. Dilution protocol using oak leaf samples. The dilution protocol was used to amplify the 297 bp fragment of chloroplastic DNA in oak leaf samples (3-step protocol, annealing at 62 °C) at different dilutions (1:100, 1:10, 1:1 and 2:1) and positive and negative controls [C+: positive control from purified DNA (Arabidopsis), C-: negative control with no template DNA]. The sample nomenclature represents the sample collection site (towns in Finland) and the sample numbering.
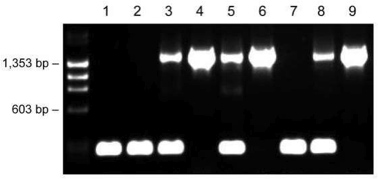
Figure 4. Genotyping of transgenic mice with one primer pair using the dilution protocol. Ear tissues of nine individual mice (lanes 1-9) were placed into 20 μl of Dilution Buffer including DNARelease Additive. After incubations at room temperature and 98 °C, 1 μl of supernatant was used as a template in the 20 μl PCR reaction. Fragment sizes: 1,500 bp (transgenic) and 200 bp (wildtype).
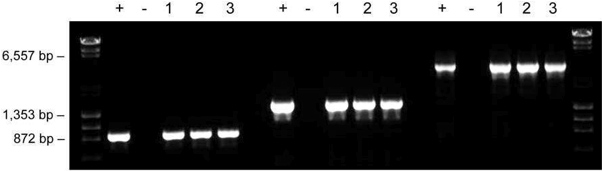
Figure 5. Samples prepared according to the dilution protocol are stable for long term storage. Samples of mouse ear tissues were incubated in 20 μl of Dilution Buffer including DNARelease Additive and subjected to repeated freezing/thawing (lane 1), stored at -20 °C for one year (lane 2), or used immediately for PCR (lane 3). The amplified fragment sizes were 900 bp, 1,500 bp and 3,200 bp.
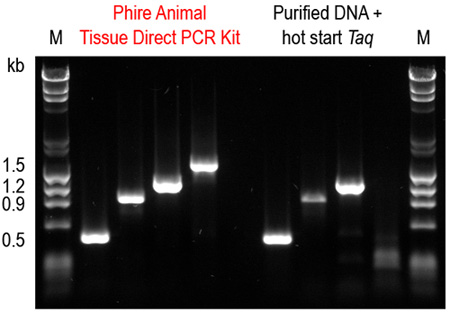
Figure 6. Phire Animal Tissue Direct PCR Kit outperforms a commercial DNA purification system combined with a hot start Taq DNA polymerase. Four amplicons (0.5-1.5 kb) were amplified from mouse ear tissues using the dilution protocol of Phire Animal Tissue Direct PCR Kit. For comparison, DNA was first purified using a commercial DNA extraction kit and the same fragments were amplified using a hot start Taq DNA polymerase according to manufacturers’ recommendations. Only with the Phire Animal Tissue Direct PCR Kit were all four amplicons successfully amplified.
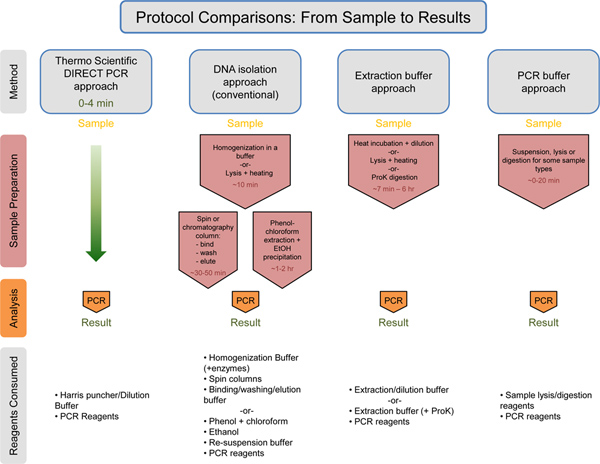
Figure 7. Universal comparison of the workflow with estimation of times/consumed reagents. Thermo Scientific Direct PCR approach is compared to the DNA isolation approach, the Extraction buffer approach, and the PCR buffer approach. The estimated time for each approach prior to the PCR step is listed in red text. The Thermo Scientific Direct PCR approach takes only 0-4 min for sample preparation prior to PCR. Below each method, the consumed reagents are listed. The Thermo Scientific Direct PCR approach uses the least amount of reagents in comparison to the other methods. Click here to view larger figure.
