1. Preparation of digitonin
- Add 500 μL of 100% ethanol to a microcentrifuge tube and place it in a heat block set at 80 °C for 10 min.
- Dissolve 2.5 mg of digitonin in 250 μL of the heated ethanol to produce a 1% stock solution of digitonin.
- To generate a working solution of 0.04% digitonin, add 40 μL of the digitonin stock solution to 960 μL of HCN buffer (50 mM Hepes pH 7.5, 150 mM NaCl, 2 mM CaCl2, 10 mM N-ethylmaleimide, and a protease inhibitor cocktail).
2. Cell intoxication and permeabilization
Our translocation assay can be applied to a range of toxins and cell lines. Below, we provide a detailed protocol for the detection of cholera toxin (CT). An overview of the procedure is provided in Figure 1.
- HeLa cells are seeded in 6-well plates containing 1 mL DMEM supplemented with 10% fetal bovine serum and antibiotic-antimycotic to achieve 1×106 cells/well after an overnight incubation at 37 °C. To generate enough cytosolic toxin for reproducible detection, triplicate wells are required for each condition.
- Following an overnight incubation, replace the culture medium with 1 mL DMEM containing 100 ng/mL of ganglioside GM1. This increases the number of binding sites for CT, as the GM1 receptor of CT will intercalate into the plasma membrane of cells exposed to a solution of GM1.
- After a 1 hr incubation at 37 °C, remove the GM1-containing medium and wash the cells twice with DMEM. Then, place the cells at 4 °C for 30 min in 1 mL DMEM containing 1 μg/mL of CT.
- Remove the toxin-containing medium, wash the cells twice with DMEM, and incubate the cells in 1 mL DMEM at 37 °C for the desired time interval(s).
- At the end of each chase period, wash the cells with PBS and incubate each well with 250 μL of 0.5 mM EDTA in PBS. Let the cells sit for 10 min at room temperature and then remove them from the 6-well plate by vigorous trituration with a p1000 pipetman. After one well of cells has been collected, combine it with the second and then third well from the same condition. Cell scrapers can also be used to collect the cells.
- Place the combined cell suspension from the three replicate wells (750 μL total volume) in a single microcentrifuge tube, and spin it in a tabletop microcentrifuge for 5 min at 5,000 x g. Cell pellets of roughly equivalent size should be obtained for all conditions.
- Discard the supernatant and resuspend the cell pellet in 100 μL of the 0.04% working solution of digitonin. Place the cell suspension on ice for 10 min.
- Spin the digitonin-permeabilized cells at 16,000 x g for 10 min in a tabletop microcentrifuge. Transfer the cytosol-containing supernatant fraction to a fresh microcentrifuge tube, and retain the organelle-containing pellet fraction.
3. Sample preparation
- Cytosol-containing supernatant fractions are diluted in HCN buffer to a final volume of 1 mL. Sample volumes of at least 1 mL are necessary to ensure air is purged from the 500 μL sample loop before injection.
- Organelle-containing pellet fractions are resuspended in 1 mL of HCN buffer containing 1% Triton X-100. Addition of detergent is necessary to release the membrane-encased pool of toxin.
- Toxin standards at concentrations of 100, 10, 1, and 0.1 ng/mL are prepared in HCN buffer.
- Parallel sets of cytosolic and organelle fractions are prepared for a control experiment to confirm the fidelity of the fractionation procedure. Fractions generated as described in Section 2 are resuspended in 20 μL of 4x sample buffer (cytosolic fraction) or 120 μL of 1x sample buffer (organelle fraction). Equivalent volumes of each fraction are resolved by sodium dodecyl sulfate polyacrylamide gel electrophoresis and probed by Western blot analysis to demonstrate the partitioning of a cytosolic protein in the supernatant fraction and a soluble, resident ER protein in the pellet fraction16-19.
4. SPR slide preparation
- The Reichert SR7000 SPR Refractometer is used for SPR experiments.
- Set a gold-plated glass slide with a self-assembled monolayer in the SPR instrument. To activate the sensor slide, perfuse a 1:1 (v:v) solution of 0.4 M EDC and 0.1 M NHS over the slide for 10 min at a flow rate of 41 μL/min. All subsequent perfusions will use the same flow rate.
- To remove the EDC:NHS activation buffer, wash the plate for 5 min with PBS containing 0.05% Tween 20 (PBST). The plate now contains reactive amide tethers due to uncapping by the EDC:NHS solution.
- Perfuse an anti-CTA antibody over the activated sensor slide at a dilution of 1:20,000 in 20 mM sodium acetate (pH 5.5) for 15 min. An initial drop in the refractive index unit (RIU) will be seen due to the pH change. This will be followed by an increase in the RIU as the antibody is captured by the amide-reactive tethers on the sensor slide.
- Remove unbound antibody from the sensor slide with a 5 minute PBST wash. The RIU signal produced by the captured antibody will plateau and stabilize, providing a new baseline signal.
- Perfuse 1 M ethanolamine (pH 8.5) over the sensor slide for 5 min. This caps and inactivates any unbound tethers left on the sensor slide.
5. SPR analysis of samples
- To establish a baseline reading, perfuse PBST over the antibody-coated sensor for 5 min.
- Perfuse an experimental sample or toxin standard over the sensor for 300 sec. Remove the ligand from the buffer and perfuse PBST over the sensor for another 200 sec.
- Following each perfusion, bound toxin is removed from the sensor by a 100 sec wash with PBST at pH 5.0. This will return the signal to its initial baseline reading, thus allowing another sample to be processed on the same sensor.
- Before loading a new sample, push a small amount of air through the sample loop to expel any residual fluid from the prior sample. Using the procedure outlined in 5.2-5.4, we have run up to 12 samples on one slide without substantial loss of the baseline signal.
- Data analysis is performed with the Scrubber 2 software, and the Igor software is used for figure preparation.
6. Representative Results
Pertussis toxin (PT) is an AB toxin that moves from the cell surface to the ER before its A chain (PTS1) enters the cytosol3, 12. As shown in Figure 2, our SPR-based translocation assay could detect PTS1 in the cytosol of intoxicated CHO cells. No signal was generated from the cytosol of unintoxicated cells, which confirmed the anti-PTS1 antibody did not cross-react with a component of the host cytosol. The cytosolic fraction from cells intoxicated in the presence of brefeldin A (BfA) also failed to produce a positive signal. BfA prevents toxin transport to the ER translocation site6-8, 12, 20-25 and, thus, A chain delivery to the cytosol. At the end of each run, bound toxin is stripped from the sensor slide. This allowed multiple samples to be screened on a single sensor slide and thereby provided a direct comparison between results obtained with different experimental conditions.
CT is another AB-type, ER-translocating toxin4. In Figure 3A, CTA1 was detected in the cytosolic fraction from CT-treated HeLa cells. This emphasized that our methodology works with multiple cell types and can be applied to any toxin for which an anti-A chain antibody is available. No signal was detected when the cytosolic fraction from CT-treated cells was perfused over an SPR sensor coated with an anti-CTB antibody (Fig. 3B), thus demonstrating that the CTA1 subunit but not the cell-binding CTB pentamer enters the cytosol. Figure 3C shows the signal from the organelle fraction is off-scale in comparison to the weaker signal from the cytosolic fraction. This was consistent with the known inefficiency of CT transport to the ER translocation site6, 7, which in turn limits the amount of toxin that can reach the cytosol. Furthermore, the organelle fraction contains CT holotoxin as well as ER-localized CTA1, so the resulting SPR signal for the organelle fraction is inflated by the additional mass of the holotoxin-associated CTB pentamer. Thus, it is not practical to plot data from the organelle and cytosolic fractions on the same sensorgram.
Our assay can monitor the time-dependent accumulation of translocated, cytosolic toxin (Fig 4). Cells were exposed to CT at 4 °C, a temperature that allows toxin binding to the plasma membrane but prevents internalization of the cell-associated toxin. After the removal of unbound toxin, the cells were warmed to 37 °C. Both toxin transport to the ER and A chain translocation to the cytosol can occur at this temperature. No toxin was detected in the cytosol 15 minutes after warming to 37 °C. This reflected the lag time required for (i) holotoxin trafficking to the ER; (ii) A/B subunit dissociation in the ER; and (iii) A chain export to the cytosol. A minor pool of cytosolic toxin was detected after 30 minutes at 37 °C, and progressively larger quantities of cytosolic toxin were detected after the 45 and 60 minute chase intervals. Even greater levels of cytosolic toxin were detected after a 5 hour chase interval17, thus demonstrating a continual, long-term delivery of cell-associated toxin to the host cytosol.
Our assay can also detect the inhibition of toxin translocation to the cytosol (Fig. 5). Cells treated with 10% dimethyl sulfoxide (DMSO), a chemical chaperone that prevents the thermal disordering of the isolated CTA1 subunit (T. Banerjee and K. Teter, unpublished observations), exhibited low levels of cytosolic CTA1 in comparison to the untreated control cells. Unfolding of the toxin A chain is a prerequisite for translocation to the cytosol16-18, so the DMSO-induced stabilization of CTA1 accordingly prevented its movement from the ER to the cytosol.
The association rate constant (ka) calculated from SPR experiments is directly proportional to the concentration of ligand in the perfusion buffer14, 15, 26. Thus, it is possible to determine the concentration of cytosolic toxin from a graph that plots the ka values for toxin standards as a function of toxin concentration. This procedure was used to quantify the DMSO-induced block of toxin translocation presented in Figure 5: the standard curve generated from known concentrations of toxin was used to calculate a cytosolic CTA1 concentration of 0.3 ng/mL for untreated cells and 0.1 ng/mL for DMSO-treated cells (Fig. 6). The inhibition of CTA1 unfolding by DMSO thus generated a 3-fold reduction in the ER-to-cytosol translocation of CTA1.
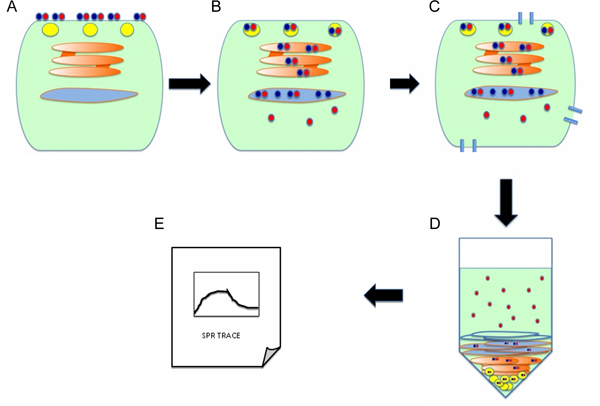
Figure 1. Protocol overview. (A) Cells are incubated with the AB toxin at 4 °C, a temperature that allows toxin binding to the cell surface but prevents toxin endocytosis. The A and B subunits of the toxin are represented by red and blue circles, respectively. (B) Unbound toxin is removed from the medium, and cells are warmed to 37 °C in order to promote endocytosis and retrograde transport of the holotoxin to the ER. Holotoxin dissociation occurs in the ER, which allows the isolated A chain to enter the cytosol by passing through a protein-conducting channel(s) in the ER membrane. (C) Cells are treated with digitonin in order to selectively permeabilize the plasma membrane. (D) Centrifugation is used to partition the cells into separate cytosolic and organelle fractions. The cytosol is squeezed out of the cell through the digitonin-generated pores and is located in the supernatant. The intact, membrane-bound organelles are found in the pellet fraction. (E) To detect the translocated pool of toxin A chain in the host cytosol, the supernatant fraction is perfused over an SPR sensor coated with an anti-A chain antibody.
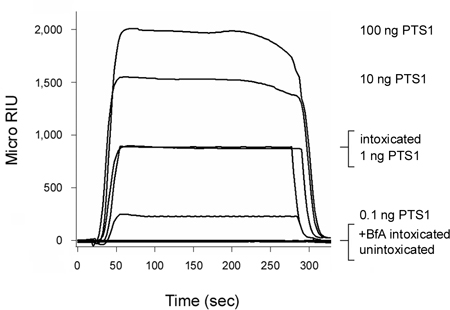
Figure 2. Detection of PTS1 translocation into the host cytosol. CHO cells were pulse-labeled at 4 °C for 30 min with 1 μg/mL of PT. The cells were then chased for 3 hr at 37 °C in toxin-free medium containing no additions (intoxicated) or 5 μg BfA/mL (+BfA intoxicated). Permeabilization of the plasma membrane with digitonin was used to partition cell extracts into separate organelle and cytosolic fractions. An SPR sensor coated with an anti-PTS1 antibody was used to detect the cytosolic pool of PTS1 from untreated or BfA-treated cells. PTS1 standards were perfused over the sensor as positive controls, while the cytosolic fraction from unintoxicated cells was perfused over the sensor slide as a negative control. At the end of each run, bound sample was stripped from the sensor slide.
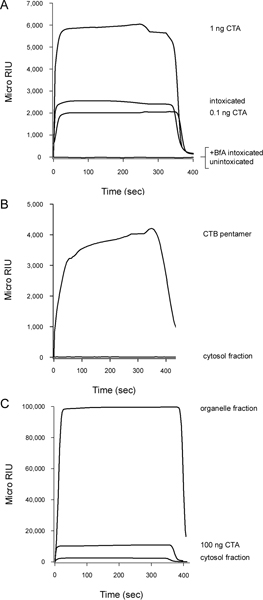
Figure 3. Detection of CTA1 translocation into the host cytosol. HeLa cells pulse-labeled at 4 °C with 1 μg/mL of CT were chased for 2 hr at 37 °C in toxin-free medium containing no additions (intoxicated) or 5 μg BfA/mL (+BfA intoxicated). Permeabilization of the plasma membrane with digitonin was used to partition cell extracts into separate organelle and cytosolic fractions. (A) The cytosolic fractions were perfused over an SPR sensor coated with an anti-CTA antibody. Known quantities of CTA were used as positive controls, while the cytosol from unintoxicated cells was used as a negative control. (B) The cytosolic fraction from cells intoxicated in the absence of BfA was perfused over an SPR sensor coated with an anti-CTB antibody. A purified CTB pentamer was perfused over the slide as a positive control. (C) The organelle fraction was solubilized with 1% Triton X-100 before perfusion over a SPR sensor coated with an anti-CTA antibody. For comparative purposes, the cytosolic fraction (1 mL final volume) from the same cell extract and a CTA standard were also perfused over the sensor. For all panels, bound sample was stripped from the sensor at the end of each run.
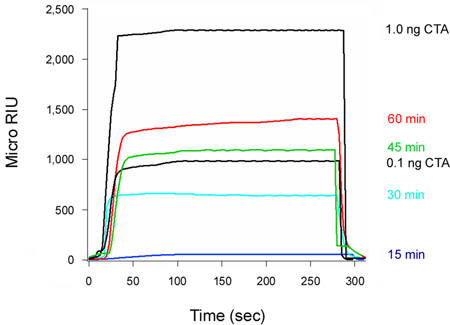
Figure 4. Kinetics of CTA1 entry into the cytosol. HeLa cells pulse-labeled at 4 °C with 1 μg/mL of CT were chased for 15, 30, 45, or 60 min at 37 °C in toxin-free medium. To detect the translocated pool of toxin, cytosolic fractions from digitonin-permeabilized cells were perfused over an SPR sensor coated with an anti-CTA antibody. CTA standards were perfused over the sensor as well. At the end of each run, bound sample was stripped from the sensor slide.
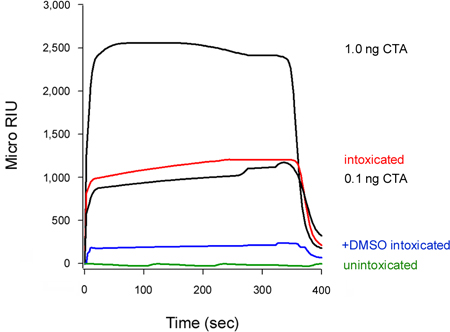
Figure 5. Inhibition of CTA1 translocation by DMSO. HeLa cells pulse-labeled at 4 °C with 1 μg/mL of CT were chased for 2 hr at 37 °C in toxin-free medium containing no additions (intoxicated) or 10% DMSO (+DMSO intoxicated). To detect the translocated pool of toxin, cytosolic fractions from digitonin-permeabilized cells were perfused over an SPR sensor coated with an anti-CTA antibody. CTA standards (100, 10, 1, and 0.1 ng/mL) were perfused over the sensor as positive controls; only the 1 and 0.1 ng/mL standards are shown for scaling purposes. The cytosol from unintoxicated cells was used as a negative control. At the end of each run, bound sample was stripped from the sensor slide.

Figure 6. Calculation of cytosolic CTA1. ka values for the CTA standards from Figure 5 were plotted as a function of toxin concentration. The resulting standard curve was used to determine, based on the ka values of the experimental samples from Figure 5, the concentration of cytosolic CTA1 in untreated and DMSO-treated cells. Toxin standards are presented as filled circles; the untreated cytosol is presented as an open square; and the DMSO-treated cytosol is presented as an open circle. The averages ± ranges of two independent experiments are shown.
