1. Generation of cell lines: pMEP4 transfection / expression
- Transfect cells with the pMEP4 expression vector and select with 100 μg of hygromycin B /ml (Roche) until all mock transfected cells have been killed off. The pMEP4 plasmid is maintained episomally so there is no need to isolate specific clones.
- Cells containing the pMEP4 vector can be induced by treatment with 10 μM CdCl2 for 16 hours. Expression and inducibility should be confirmed on a small scale before amplification of the cell lines. The TAP-tagged protein can be readily detected as the protein G domains bind to antibodies from almost all species. For large-scale purifications typically 10 confluent 175 cm2 flasks of cells are required. This equates to approximately 2 X108 expressing cells.
2. Cell lysate preparation
- Scrape cells into PBS and combine into a single 50 ml tube. Spin 1200X g for 5 minutes (this should result in 2-3 ml of packed cells to start with, which reduces to about 1.5 ml after washing).
- Wash the cells three times in ice-cold PBS (50 mls each time).
- Lyse cells in 5 ml lysis buffer (50 mM Tris-HCl (pH 7.5), 125 mM NaCl, 5 % Glycerol, 0.2 % NP-40, 1.5 mM MgCl2, 25 mM NaF, 1 mM Na3VO4 and protease inhibitor). Note: NaF, Na3VO4 and protease inhibitors should be added fresh. Pipette up and down 10 times before leaving for 5 minutes on ice. Syringe up and down 5-10 times using a blunt needle before leaving again on ice for 5 minutes on ice. Repeat syringing and leave for a further 5 minutes on ice.
- Freeze-thaw the lysate twice (liquid N2 / or dry ice and ethanol). Do not allow sample to reach greater than 4°C. Note: you can store this sample at -80°C until you have time to process.
- Aliquot sample into 1.5 ml tubes and remove unlysed cells and debris by centrifugation (10 minutes at 4°C, 16,000X g).
- Recover supernatant, combine in a 15 ml falcon tube and (optionally) pass through a 0.45 μm filter before taking a 20 μl sample for protein yield quantification. Remove a further 50 μl aliquot for subsequent analysis (Sample 1).
3. Binding to Rabbit IgG-agarose
(Note: All spins should be carried out at 1200X g in a refrigerated centrifuge at 4°C for 1 minute, unless stated otherwise)
- Gently resuspend the rabbit IgG-agarose solution (Sigma-Aldrich) by swirling bottle. Take 380 μl of IgG agarose (using a cut 1ml pipette tip) and remove shipping/preservative solution by centrifugation for 1 minute. Wash the agarose three times in chilled lysis buffer (4°C) using centrifugation to clarify. The final yield of agarose should be about 250 μl of packed beads.
- Add the cleared cell lysate from 2.6 (in a 15 ml falcon tube) to the washed rabbit-IgG agarose and incubate for 3 hours (or overnight) at 4°C using a rotating mixer.
- Spin down the agarose beads for 5 minutes at 4°C and remove the supernatant for subsequent analysis (Sample 2). Note: The TAP tagged protein should now be associated with the beads.
4. TEV protease cleavage
(Note: All spins should be carried out at 1200X g in a refrigerated centrifuge at 4oC for 1 minute, unless stated otherwise)
- Wash the rabbit IgG agarose beads three times in chilled lysis buffer (4°C) using centrifugation to clarify (note: the lysis buffer should not contain protease inhibitors). Care should be taken not to remove or lose any beads during the washing steps.
- Wash the beads a further two times with TEV-protease cleavage buffer (10 mM Tris-HCl (pH 7.5), 100 mM NaCl, and 0.2 % NP-40) using centrifugation to clarify. After the last wash carefully remove all liquid from the beads.
- Prepare a TEV protease cleavage mix; for each sample include 467.5 μl H2O, 25 μl 20x TEV Buffer (Invitrogen), 5 μl 0.1M DTT and 2.5 μl (25 U) TEV Protease (Invitrogen). Add 500 μl of this TEV cleavage mix to each sample of packed beads and transfer the entire mix to a 1.7 ml pre-lubricated tube (Costar). Prior to incubation remove a 30 μl aliquot for subsequent analysis (Sample 3).
- Incubate the TEV protease reaction overnight at 4°C using a rotating mixer. Note that shorter incubations times may be used depending on the nature of the bait protein and the accessibility of the TEV protease cleavage site but the minimal incubation time should be empirically determined.
5. Binding to Ultralink Immobilized Streptavidin Plus beads
(Note: All spins should be carried out at 1200X g in a refrigerated centrifuge at 4°C for 1 minute, unless stated otherwise)
- Centrifuge the TEV cleavage reaction containing the rabbit IgG agarose beads for 5 minutes at 1200X g (4°C). Remove a 20 μl aliquot of the clarified supernatant for analysis (Sample 4)
- Transfer the remaining supernatant (roughly 480 μl) to a fresh 1.5 ml pre-lubricated tube (Costar) and leave this on ice. Do not throw away this supernatant – this contains your TEV cleaved bait protein and any associated binding proteins.
- Add 500 μl of chilled lysis buffer (Section 2.3) to the remaining rabbit IgG agarose beads and resuspend. Centrifuge this mixture to clarify the beads before removing the supernatant and combining it with that from step 5.2. Leave this tube on ice (it should now contain ~980 μl of sample).
- Retain the rabbit IgG agarose beads for subsequent analysis (Sample 5). Note: if you run this sample on an SDS-PAGE gel (or western blot) it will give a lot of background due to the presence of the IgG heavy and light chains.
- Meanwhile gently resuspend the Ultralink Immobilized Streptavidin Plus agarose beads (Pierce) by swirling the bottle. Using a 200 μl pipette tip with the end cut, aliquot 70 μl of streptavidin beads per sample into pre-lubricated 1.5 ml tubes and remove shipping/preservative solution by centrifugation. Note: these beads are very small and so “duck-billed” or flat/narrow-ended tips can be used to minimize bead loss during washing.
- Wash the streptavidin beads three times in chilled lysis buffer (4°C) using centrifugation to clarify. This should leave approximately 45-50 μl of packed beads per tube.
- Transfer the TEV-protease cleavage supernatant (i.e. the 980 μl of sample from step 5.3) to the tube containing the washed streptavidin beads and incubate for 3 hours (or overnight) at 4°C using a rotating mixer.
- Spin down the streptavidin beads for 5 minutes at 4°C and remove the supernatant for subsequent analysis (Sample 6). Wash the streptavidin beads three times in chilled lysis buffer (4°C) using centrifugation to clarify. After the final wash, carefully remove all remaining wash buffer.
6. Biotin elution of the streptavidin binding peptide and bait protein
(Note: All spins should be carried out at 1200X g in a refrigerated centrifuge at 4°C for 1 minute, unless stated otherwise)
- Elute tagged proteins from the streptavidin beads by adding 500 μl of PBS:Biotin (1 mM D-biotin) and incubating at 4°C for 3 hours (or overnight) using a rotating mixer.
- Spin down the streptavidin beads for 5 minutes at 4°C and remove the supernatant to a 1.5 ml pre-lubricated tube (Note: this is your final sample/ elution (Sample 7).
- Add another 500 μl Biotin:PBS to the remaining streptavidin beads and incubate at 4°C for 2-3 hours (or overnight) using a rotating mixer.
- Repeat step 6.2 and combine the two elutions (Sample 7). This final elution can be stored at -80°C.
- To assess the efficiency of the biotin elution, freeze the remaining streptavidin beads and boil later in SDS sample buffer (Sample 8) (recommendations: LaneMarker 5x reducing sample buffer from Fisher).
7. Protein concentration
- The final eluate (Sample 7) needs to be concentrated prior to analysis because of the low protein concentration and relatively high volume. This can achieved using low molecular weight (<5 KDa) Vivaspin spin columns (Vivascience). Aim to concentrate the volume down to less than 100 μl. This final sample can be boiled and stored in protein sample buffer (recommendations: LaneMarker 5x reducing sample buffer from Fisher).
8. Analysis
- Samples 1-8 can be analyzed by western blot to determine the efficiency of pull down. Note: Most secondary antibodies will cross-react with the Protein G domains of the fusion protein but this is removed after TEV protease cleavage.
- Sample 7 can be analyzed by 1D or 2D SDS-PAGE. Staining can be performed using silver or Coomassie (recommendations: SilverQuest Silver staining and Colloidal Blue Coomassie staining kits from Invitrogen). Protein identification can be performed using mass spectrometry.
9. Representative Results
An example of 1D SDS-PAGE analysis of the final elution (Sample 7) from this protocol to identify binding partners of TAP tagged eIF4E is provided in Figure 2. This representative gel reflects the complex and abundant nature of eIF4E interactions with other proteins in the cell. Comparison with the negative control also shown in Figure 2, generated from a cell line expressing only the TAP tag, illustrates the specificity of this eIF4E baited pull-down. In this instance 15% of the concentrated final elution (Sample 7) was analyzed by 1D SDS-PAGE using commercially available pre-cast gradient gels before being stained using the Silverquest silver staining kit from Invitrogen.
Typically 50-85% of the remaining concentrated final elution (Sample 7) is analyzed with Colloidal Coomassie stain (Invitrogen). Samples (gel slices/bands) from the entire lane were then extracted and analysed by mass-spectrometry.
Proteins identified in the eIF4E pull-down were filtered against the binding partners from the negative control to identify non-specific binding partners. The final proteins identified using this process of TAP tagging eIF4E can be seen in Figure 2, which is representative of the normal proteins identified using this technique. In addition, a number of the eIF3 subunits were also identified (data not shown).
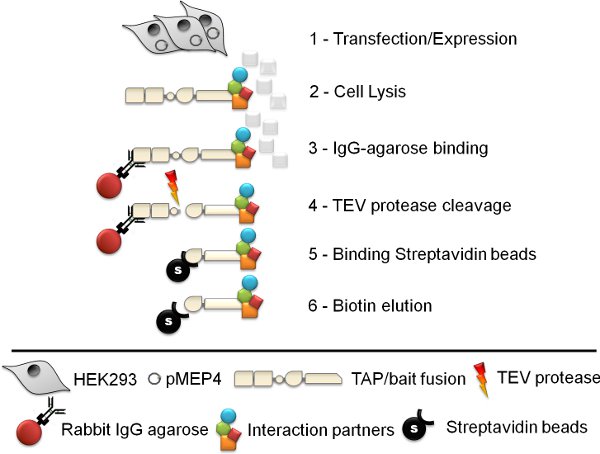
Figure 1. Schematic of the tandem affinity purification procedure. The six step tandem affinity purification (TAP) protocol involves cell line generation, cell lysis, Rabbit IgG agarose immuno-precipitation, TEV protease cleavage, streptavidin bead affinity purification and finally biotin elution.
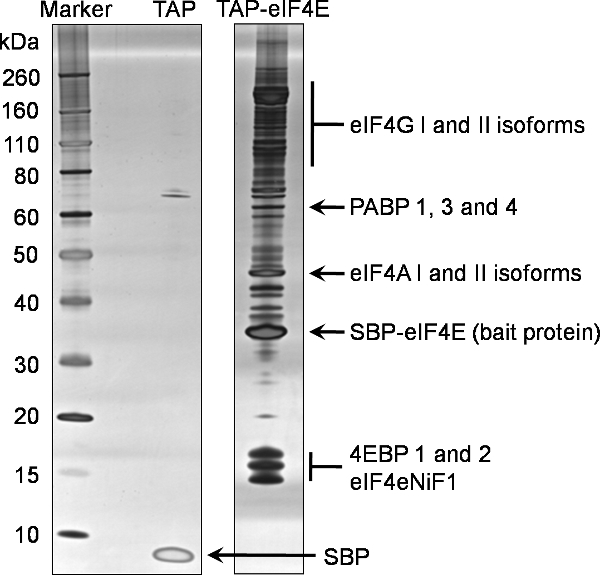
Figure 2. Tandem affinity purification of the murine eIF4E protein. Interacting partners of the N terminally TAP tagged eIF4E were purified from eukaryotic HEK293 cells using the attached protocol. A 20% fraction of the final elution (Sample 7) was analysed by SDS-PAGE on a pre-cast 4-12% gradient gel (TAP-eIF4E lane). An equivalent analysis was performed for the tag alone (TAP lane). Proteins were identified using silver staining. Proteins subsequently identified by mass spectrometry of the same sample are highlighted on this gel.
Abbreviations: eIF4G; eukaryotic translation initiation factor 4 gamma, PABP; polyA binding protein, eIF4A: eukaryotic translation initiation factor 4 alpha, SBP-eIF4e; the remaining TAP bait protein containing the streptavidin binding peptide fused to the eukaryotic translation initiation factor 4E, 4EBPs; eukaryotic translation initiation factor 4E binding proteins, eIF4eNiF1l eukaryotic translation initiation factor 4E nuclear import factor 1, SBP; the remaining streptavidin binding peptide from the TEV unfused TAP peptide.
