Safety
The protocol described below utilizes ATP labelled with radioactive 32P at the γ phosphate position to follow the kinase activity of LRRK2. It is based on the standard protocols used in our laboratory, and so with regard to many of the steps in the process such as running the gels, western blotting et cetera the materials and precise conditions should be taken as a guide as the equipment and protocols used for these processes varies from laboratory to laboratory. Compounds containing isotopes that emit ionising radiation are potentially harmful to human health and strict licensing and regulations at an institutional and national level control their use. The experiments in this protocol were carried out following training in open source radiation use at University College London and following the good laboratory practise guidelines provided by the safety services at the college (guidelines available at http://www.ucl.ac.uk/estates/safetynet/training/). Use of open source radiation should not be attempted prior to appropriate training and regulatory approval. The regulatory body responsible for open source radiation in laboratory research varies from country to country. Examples of these are: in the United Kingdom, the Health and Safety Executive (http://www.hse.gov.uk/radiation/ionising/index.htm), in the United States the Nuclear Regulatory Commission ( http://www.nrc.gov/materials/miau/regs-guides-comm.html), in Canada the Canadian Nuclear Safety Commission (http://nuclearsafety.gc.ca/eng/), and in Germany Das Bundesamt für Strahlenschutz (http://www.bfs.de/de/bfs). Users in other countries should confirm local rules, regulations and licensing authorities with their radiation safety officer. Safety precautions relevant to this protocol have been noted in the text, highlighted with the radioactive trefoil symbol ( ).
).
1. Preparing the kinase reactions
All reaction mixtures prepared in 1.5ml sample tubes with screw caps containing an O ring to prevent spread of radioactivity.
- Thaw protein on ice – LRRK2 wild type, D1994A, G2019S.
- make up reaction on ice – 10nM LRRK2, 0.5μg/μl MBP, 5ul of 10x kinase buffer, made up to 50μl with water.
2. Running the assay
All steps utilizing 32P ATP should take place in designated radiation areas.
Suitable personal protective equipment should be worn – under standard operating procedure in our laboratory these include lab coat, double gloves and protective goggles.
Samples containing 32P ATP should be shielded from users by 6mm Perspex screens to minimize exposure.
Where applicable, personal monitoring devices should be used – within UCL, any certified open source radiation user must have a film badge to monitor radiation exposure during experiments.
All experimental surfaces should be assessed for radioactive contamination before and after use using a Geiger counter.
All potentially contaminated consumables should be disposed of in strict adherence to institutional guidelines for radioactive waste disposal.
- Prior to starting assay, set heating blocks to 30°C and 100°C respectively.
- Remove 32P ATP from -20 freezer (note that storage conditions for 32P ATP may vary depending on supplier or type of radionucleotides used). scan outside of container prior to use, thaw behind perspex screen.
- With reactions on ice, add 1μl of 32P ATP to each along with 10μM of cold ATP.
- Mix well with pipette. Pulse centrifuge to bring liquid to bottom of tube, minimizing risk of contamination.
- Remove 15μl aliquot for zero time point and terminate reaction in aliquot by addition of 5μl of 4x SDS sample buffer and denaturation at 100°C for 10 minutes. Pulse centrifuge to bring liquid to bottom of tube, minimizing risk of contamination.
- Remaining sample placed in heating block and incubated at 30°C for 60 minutes.
- 15μl removed at 60 minute time point and reaction terminated by addition of 5μl of 4x SDS sample buffer and denaturation at 100°C for 10 minutes. Pulse centrifuge to bring liquid to bottom of tube, minimizing risk of contamination.
3. Immunoblotting the samples and analysing results
- Samples run on SDS-PAGE
- 10 well 4-12% Bis-tris polyacrilamide gel prepared for electrophoresis using MOPS running buffer.
- 20μl of each sample loaded onto gel along with 7μl of sharpstain protein standard ladder.
- Gel run at 160v for 90 minutes, or until dye front has reached the end of the gel. All liquid in contact with radioisotopes should be treated as radioactive waste and disposed of as per institutional guidelines. UCL regulations state that liquid radioactive waste should be discarded by pouring down designated radioactive disposal sinks with copious water.
- Protein transferred to PVDF membrane via western blot.
- Transfer buffer prepared, 1x Tris Glycine plus 20% Methanol. PVDF Membrane and filter paper cut to correct size for gel and activated with glacial methanol in the case of the membrane or pre-wet with transfer buffer in the case of the filter paper. The membrane should be pre-labelled with ballpoint ink to allow identification of orientation. Gel removed from plastic casing and excess acrylamide removed and disposed of as radioactive waste.
- The gel should be formed into a sandwich with the membrane and filter paper, ensuring that no bubbles exist between the membrane and gel, and then arranged in the western blot apparatus with the membrane between the gel and the anode.
- Transfer carried out at 25V for 16 hours.
- Transfer buffer prepared, 1x Tris Glycine plus 20% Methanol. PVDF Membrane and filter paper cut to correct size for gel and activated with glacial methanol in the case of the membrane or pre-wet with transfer buffer in the case of the filter paper. The membrane should be pre-labelled with ballpoint ink to allow identification of orientation.
- Following protein transfer to PVDF, the membrane should be dried at room temperature. Finally, the dry membrane should be isolated between cellulose acetate sheets and exposed to either a phosphor screen or x-ray film to allow detection of radiolabelled protein. Exposure time can run from several hours to over a week depending on the specific activity of the radioisotope used and the enzyme kinetics of the reaction.
- Phosphoscreen scanned/film processed. If using a phosphor screen, the image should be saved as a high-resolution TIFF or bitmap file, if using film then the resultant transparency should be scanned using a desktop scanner and saved as a high-resolution TIFF or bitmap file. The image can then be analyzed in ImageJ, a freeware program available on the National Institutes of Health website (http://rsbweb.nih.gov/ij/).
4. Representative Results
Figure 1 shows representative results for an assay carried out using wild type, G2019S and D1994A LRRK2 with Myelin Basic Protein as a generic phosphate acceptor substrate. Autophosphorylation of LRRK2 is visible at ≈ 200kDa, with multiple bands representing phosphorylated MBP visible from 20-40kDa. Note the absence of autophosphorylation in the D1994A (kinase dead) lane, and increased phosphorylation due to the G2019S mutation. Note also residual phosphorylation of MBP in the kinase dead lane. This may be due to incomplete ablation of the kinase activity of LRRK2 by the D1994A mutant or reflect the presence of trace contaminating kinases in the reaction.
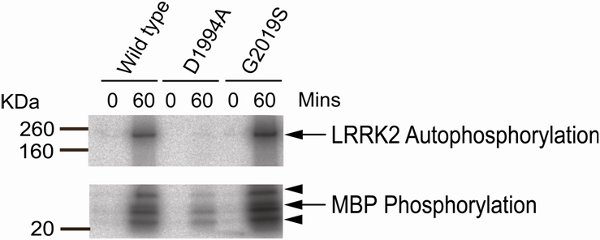
Figure 1.
