Colonic mucosal biopsies were collected from patients with CD undergoing routine colonoscopy as part of the standard of care. Ethical approval for the use of patient tissue samples and the generation of colonic organoid lines from these samples was obtained from the Clinical Research Ethics Committee of the Cork Teaching Hospitals (CREC). Written informed consent was obtained from all patients in agreement with the Declaration of Helsinki. All tissue culture work with patient biopsies and colonoids must be performed inside a biosafety cabinet following BSL2 safety protocols. Ensure all plastic wear is sterile before use. See the Table of Materials for details related to all materials, reagents, instruments, and software used in this protocol.
The protocols our group uses for crypt isolation and organoid culture were adapted from established methods14,15,16 and have been published previously9,10,17. For the following protocol colonoids were cultured using Organoid Proliferation Media (Table 1). Colonoids grown using Organoid Proliferation Media are undifferentiated and enriched for colonic stem cells. The primary component of Organoid Proliferation Media is 50% L-WRN conditioned media, which contains the intestinal stem cell niche growth factors Wnt-3A (W), R-spondin 3 (R), and Noggin (N)15. Organoid Proliferation Media is prepared by combining L-WRN conditioned media and Serum-free Media 1:1, followed by supplementation with nicotinamide and chemical inhibitors (Table 1).
1. Colonic crypt isolation and colonoid culture
- Preincubate a 48-well microtiter plate at 37 °C, 5% CO2 for a minimum of 72 h before seeding with crypts.
NOTE: Preincubating the plate accelerates the polymerization of the basement membrane extract (BME) during seeding. - Defrost the BME on ice at 4 °C the evening before the isolation of crypts.
- Collect the colonic biopsies in a sample collection tube containing 15 mL of Biopsy Collection Medium (Table 1) and store at 4 °C until ready for processing.
- Carefully remove as much Biopsy Collection Medium as possible using a pipette. Wash the biopsies by adding 15 mL of ice-cold DPBS supplemented with 2.5 µg/mL amphotericin B and 100 µg/mL gentamicin to the sample tube. Shake the sample tube vigorously to dissociate any mucous or debris from the biopsies. Let the biopsies settle by gravity and carefully remove as much DPBS as possible using a pipette.
- Repeat washing procedure from step 1.4 two times (2x).
- Add 10 mL of enzyme-free cell dissociation reagent supplemented with 2.5 µg/mL amphotericin B and 200 µg/mL gentamicin to the sample tube and incubate for 15 min at room temperature with rocking at 30 rpm.
- After the incubation, shake the sample tube side to side by hand vigorously to release the colonic crypts. Inspect the tube using a low-powered light microscope and look for the released crypts and crypt fragments in suspension. If not visible, shake the tube and check again; repeat until the crypts are seen in suspension.
- Attach a 70 µm cell strainer to a 50 mL tube and filter the crypt suspension through the strainer. Add 10 mL of ice-cold Organoid Wash Media (Table 1) to the empty sample tube, remove the media, and pass it through the cell strainer.
- Transfer the filtered crypts into two 15 mL tubes (10 mL per tube) and centrifuge at 4 °C for 5 min at 150 × g.
- Remove the supernatant carefully from each 15 mL tube, resuspend the crypt pellets in 500 µL of ice-cold Organoid Wash Media, transfer the crypt solution from both 15 mL tubes to a single 1.5 mL microcentrifuge tube, and centrifuge at 4 °C for 3 min at 400 × g.
- Carefully remove the supernatant from the microcentrifuge tube and resuspend the crypt pellet in 70 µL of BME (20 µL per well and 10 µL of extra dead volume).
- Using the preincubated 48-well plate (step 1.1), seed 20 µL of BME/crypt suspension into the center of each well (1 BME dome per well). Smoothly and steadily invert the plate and incubate at 37 °C, 5% CO2 for 20 min.
NOTE: Inverting the plate prevents cells from adhering to the plastic surface of the well and ensures their distribution within the BME. - Remove the plate from the incubator. Ensure the BME has fully polymerized and then overlay the domes with 350 µL of prewarmed Organoid Proliferation Media supplemented with 100 µg/mL of a broad-range antimicrobial reagent. Incubate the plate at 37 °C, 5% CO2.
NOTE: The broad-range antimicrobial reagent is to prevent contamination from mucosal microbes associated with the colonic biopsies. It is only required for the first week of culture following crypt isolation. - Change the media 2-3x in a week using prewarmed Organoid Proliferation Media, supplementing with the broad range antimicrobial reagent for the first week of culture; following this period, remove the reagent.
- Once the colonoid culture is fully established (1-2 weeks post isolation), dissociate the colonoids using the enzymatic dissociation reagent supplemented with 10 µM of the ROCK-I/II inhibitor Y-27632 (see section 2 for full details on colonoid dissociation).
NOTE: For the first two passages (P0-1, P1-2), colonoids should be expanded using a 1:1/2 ratio; after P2, colonoids can be passaged using a 1:3/4 ratio. - Seed and maintain the colonoids following steps 1.11-1.13 (do not supplement Organoid Proliferation Media with the broad range antimicrobial reagent).
2. Preparation of colonoids for cell death assay
NOTE: The cell death assay protocol takes 4 days to complete (Figure 1A).
- Expand the colonoids using a 48-well plate format, seeding 20 µL of BME/crypt suspension per well, incubating at 37 °C, 5% CO2, and changing the medium 2-3x a week (350 µL per well).
- Passage the colonoids approximately 1 week prior to harvesting for the cell death assay. Before harvesting the colonoids, ensure they are propagated to a high density (Figure 1Bi), that they are approximately 25-50 µm in diameter, and are actively proliferating.
NOTE: We have used colonoids for this assay from passage 3 to passage 14 with consistent results. However, it has been demonstrated that the transcriptional response of colonoids to cytokines can change depending on culture duration18. On this basis, we recommend not to use colonoids for this assay after passage 15. - Preincubate 96-well microtiter plates at 37 °C, 5% CO2 for a minimum of 72 h before seeding with colonoid cells.
- Defrost the BME on ice at 4 °C the evening before starting the experiment.
- Prepare a sufficient volume of the enzymatic dissociation reagent (500 µL per well of colonoids) by supplementing with 10 µM of the ROCK-I/II inhibitor Y-27632. Gently remove the medium from the wells containing colonoids; pipette from the edge of the well to avoid damaging the colonoid dome. Add 300 µL of enzymatic dissociation reagent to each well.
- For each well, break apart the colonoid dome by scraping the surface of the well with the tip of a P1000 pipette and pipetting the cell suspension up and down; try to avoid generating air bubbles. Collect the cell suspension in a 15 mL tube.
NOTE: This 15 mL tube will be used to collect colonoids from 10 wells of the 48-well plate. - Wash the same well with another 200 µL of the enzymatic dissociation reagent to ensure that all colonoid material is collected and transferred to the same 15 mL tube. Repeat this process for each well of expanded colonoids being harvested and collect them in the same single 15 mL tube.
NOTE: For efficient dissociation, a maximum of 10 wells of colonoids should be collected per tube. If collecting >10 wells, split the collected colonoids equally across multiple 15 mL tubes.
- For each well, break apart the colonoid dome by scraping the surface of the well with the tip of a P1000 pipette and pipetting the cell suspension up and down; try to avoid generating air bubbles. Collect the cell suspension in a 15 mL tube.
- Incubate the 15 mL tube with collected colonoids from step 2.5.2 in a water bath at 37 °C for 5 min. Following the incubation, centrifuge the tube for 3 min at 400 × g. Gently remove the supernatant, leaving approximately 1.2 mL behind in the tube.
NOTE: The next step requires the physical dissociation of colonoids using rapid pipetting. For this to be effective, you must have a small volume of cell suspension in the tube – the 1.2 mL left in the 15 mL tube is sufficient for this purpose. - Resuspend the colonoid pellet in the remaining 1.2 mL of the enzymatic dissociation reagent. Using a P1000 pipette set to 1,000 µL, place the tip of the pipette into the suspension, holding it just above the bottom of the 15 mL tube, then rapidly pipette the suspension in and out of the tip. To rapidly pipette, quickly depress the plunger button to the first stop, release the button until it is approximately halfway to the top position, and repeat. Rapidly pipette for approximately 10 s (30-40 depressions), then resuspend the suspension fully and repeat the pipetting. Perform 2-3 rounds of rapid pipetting.
- Check the sample under the microscope to confirm that there are no whole colonoids left and that the majority of colonoid fragments are approximately 30-40 µm in size (Figure 1Bii).
- If the sample requires further dissociation, incubate in the water bath at 37 °C for another 3 min, repeat the pipetting technique in step 2.7, and check the sample under the microscope. Repeat this process until the majority of the colonoid fragments are of the optimal size.
NOTE: Be careful not to dissociate the colonoids too much as this will result in excessive cell death, low plating efficiency, and undersized colonoids. - Add 10 mL of ice-cold Organoid Wash Media (Table 1) to the 15 mL tube. Centrifuge the tube for 3 min at 400 × g, remove the supernatant, resuspend in 1 mL of ice-cold Organoid Wash Media, and transfer to a 1.5 mL microcentrifuge tube (referred to as the colonoid fragment tube).
- Mix the contents of the colonoid fragment tube by pipetting and take a 50 µL sample; transfer this 50 µL sample to a new 1.5 mL microcentrifuge tube (referred to as the cell count tube). Store the colonoid fragment tube on ice from this point until the completion of step 2.15.
- Centrifuge the cell count tube for 3 min at 400 × g, remove the supernatant, and resuspend in 500 µL of enzymatic dissociation reagent supplemented with 10 µM of the ROCK-I/II inhibitor Y-27632.
- Incubate the single cell count tube in the water bath at 37 °C for 5 min. Using a P1000 pipette set to 400 µL, pipette the sample as described in step 2.7 and check the sample under the microscope to ensure there is a single-cell suspension. If not, repeat this process until the colonoids are fully dissociated into a single-cell suspension.
- Add 1 mL of Organoid Wash Media to the single cell count tube, centrifuge for 3 min at 400 × g and carefully remove the supernatant. Resuspend the cells in 50 µL of Organoid Wash Media and then add 50 µL of trypan blue.
- Count the cells using a hemocytometer. Calculate the number of cells in the 50 µL sample and use this to calculate the concentration of cells in the colonoid fragment tube.
- Calculate the volume of cell suspension required for the experiment where 0.5 × 104 colonoid cells will be seeded per well of a 96-well microtiter plate. Add approximately 15% extra to this calculated volume to account for dead volume. Transfer this total volume from the colonoid fragment tube (step 2.11) to a new 1.5 mL microcentrifuge tube.
- Centrifuge the new 1.5 mL microcentrifuge tube containing colonoid fragments for 3 min at 400 × g, remove the supernatant, and resuspend in BME (use 10 µL of BME per 0.5 × 104 cells).
NOTE: Due to the physical characteristics of the BME (high viscosity, temperature-dependent polymerization), a significant quantity of material can be lost through pipetting (dead volume).
- Centrifuge the new 1.5 mL microcentrifuge tube containing colonoid fragments for 3 min at 400 × g, remove the supernatant, and resuspend in BME (use 10 µL of BME per 0.5 × 104 cells).
- Set up the tube or reservoir containing the colonoid/BME suspension on ice in a sterile container inside the biosafety cabinet.
NOTE: Keeping the cells on ice while seeding prevents the cell substrate from polymerizing prematurely. - Using a preincubated 96-well microtiter plate (from step 2.3), reverse pipette 10 µL of the colonoid/BME solution per well. Be sure to position the tip just above the surface of the well and pipette into the center to avoid hitting the wall of the well. Mix the colonoid/BME suspension regularly to prevent uneven seeding.
NOTE: Do not seed colonoids in the outer edge wells of the microtiter plate.- To reverse pipette:
- Set the pipette and then press the plunger button past the first stop to the second stop.
- Holding this position, immerse the tip in the colonoid/BME suspension and slowly release the plunger to the top.
- Dispense the suspension by pressing the plunger button gently and steadily to the first stop. If seeding more wells, hold this position and repeat steps 2.18.1.2-2.18.1.3.
- Once finished, expel the small amount of remaining suspension by pressing the plunger button to the second stop.
NOTE: We recommend using the reverse pipetting technique due to the high viscosity of the BME.
- To reverse pipette:
- Smoothly and steadily invert the plate and incubate at 37 °C, 5% CO2 for 20 min.
- Remove the plate from the incubator. Ensure the BME has fully polymerized and then, overlay the domes with 200 µL of prewarmed Organoid Proliferation Media. If using a 96-well microtiter plate with a surrounding moat (that reduces evaporation), fill each reservoir with 2 mL of Organoid Wash Media.
- Incubate the colonoids at 37 °C, 5% CO2 for 3 days and inspect microscopically once a day to ensure the colonoids have recovered and are proliferating.
3. Colonoid treatments for cell death assay
- Prepare a 2.5 µM solution of fluorescent cell death dye (SYTOX Green Nucleic Acid Stain) and DMSO solution by adding fluorescent cell death dye or DMSO to prewarmed Organoid Proliferation Media (Table 2).
NOTE: Protect the fluorescent cell death dye stock and diluted solution from light. SYTOX Green Nucleic Acid Stain is solubilized in DMSO; the DMSO solution is used to prepare the No Dye condition to control for solvent effects. - Use the 2.5 µM solution of fluorescent cell death dye and DMSO solution from step 3.1 to prepare treatments as shown in Table 2.
- Gently remove the medium from the 96-well microtiter plate seeded with colonoids by tilting the plate and pipetting from the edge of the wells; then, add 200 µL of treatment medium per well. Be sure to have extra PBS/BSA control wells for the Max Toxicity condition(s) (see Table 2 for the plate map).
- Incubate the colonoids at 37 °C, 5% CO2 for the required treatment time points until ready for imaging.
- At least 2 h before imaging, prepare a 10% v/v solution of Triton-X 100 in sterile cell-culture grade water and add 22 µL of the 10% Triton-X 100 directly to the medium of the control wells for the Max Toxicity condition(s) to a final concentration of 1% v/v 1 h before imaging.
NOTE: Using a 10% solution reduces pipetting errors that can occur due to the high viscosity of the surfactant.
4. Image acquisition
- Remove the 96-well microtiter plate seeded with colonoids from the incubator and transfer to the stage of a digital inverted epifluorescence microscope. Allow the plate to come to room temperature.
- Confirm that the Max Toxicity condition colonoids are fully lysed by examining them under the microscope (Figure 1Biii).
- Select a suitable objective, such as a long working distance 40x fluorescence objective. Optimize the imaging settings of the microscope before beginning.
- Using the transmission channel, focus on a colonoid with SYTOX-positive cells, switch to the Green Fluorescent Protein (GFP)
channel (488 nm), and adjust the light intensity and exposure time to maximize the fluorescent signal while minimizing the background. First, try a low light intensity, then gradually increase the exposure time; if the length of exposure is impractical, then increase the light intensity slightly.
NOTE: Increasing the exposure time instead of the light intensity will reduce phototoxicity and photobleaching of the samples19. Only expose the samples to fluorescent light when necessary.
- Using the transmission channel, focus on a colonoid with SYTOX-positive cells, switch to the Green Fluorescent Protein (GFP)
- Using the optimized imaging settings for the GFP channel, observe the No Dye and Max Toxicity conditions to ensure the samples are not over- or underexposed. Once finalized, keep the imaging settings consistent between conditions.
- Acquire images using a random sampling approach: do this by selecting fields of view (FOVs) that follow a fixed grid pattern that covers the colonoid dome. Acquire images of colonoids from a minimum of 10 FOVs. Ensure the central plane of the colonoid is in focus and acquire images in both transmission and GFP channels.
- Apply the following exclusion criteria.
- Do not acquire images if there are no colonoids present in the FOV.
- Do not acquire images if there are only colonoids that are overlapping in the same focal plane present in the FOV (Figure 1Biv).
- Do not acquire images if there is only colonoid debris present in the FOV (Figure 1Bv). Do not include colonoid debris in the analysis.
- Apply the following exclusion criteria.
- Save images in portable network graphic format and export.
- If imaging additional time points, return the 96-well microtiter plate with colonoids to the incubator at 37 °C, 5% CO2.
5. Image analysis
- Open Fiji ImageJ and import the image dataset by dragging and dropping the files onto the ImageJ toolbar or navigating to File | Open and selecting the files. Once open, combine the files into an image stack by clicking Image| Stacks| Images to Stack. Convert the image stack to 8-bit file format by clicking Image| Type| 8 -bit.
- For each image set, click the Freehand Selections tool on the ImageJ toolbar and manually select the region of interest (ROI) on the transmission image using the computer mouse; the ROI is the perimeter of the colonoid. Then, toggle through the image stack to the corresponding GFP channel image.
- Click Analyze| Set Measurements; in the Set Measurements dialog window, tick Mean Grey Value and leave all other boxes unticked. With the GFP image selected, click Analyze | Measure. Repeat this analysis for every colonoid in the image stack. Once the dataset is analyzed, copy all the data in the 結果 window and paste them into a spreadsheet software application.
6. % max toxicity calculation
- Calculate the mean of the technical replicate Mean Grey Values (MGV) for each condition using equation (1).
 (mean of treatment a) =
(mean of treatment a) =  (1)
(1) - Express the mean of each condition as a percentage relative to the mean of the Max Toxicity condition (MT) using equation (2).
%MTa = (2)
(2)
7. Calculating the CPI
- Normalize (NORM) the data as follows, using the mean values calculated in step 6.1, subtract the mean of the Untreated condition (UT) from each treatment condition and the Max Toxicity (MT) condition (to remove the background cell death that occurs independent of cytokine treatment). Then, divide each treatment condition by the background-subtracted mean of the Max Toxicity (MT) condition as shown in equation (3).
NORMa = (3)
(3) - Subtract the normalized values calculated in step 7.1 from 1; the resulting values represent cell viability (V) after treatment (as in equation (4) below).
Va = 1 −NORMa (4) - Calculate the coefficient of perturbagen interaction (CPI) using equation (5):
CPI = (5)
(5)
Where a denotes the first treatment; b denotes the second treatment; and ab is the combination treatment. CPI values indicate synergistic (<1), additive (=1), or antagonistic (>1) relationships.
Using this protocol, we demonstrated how CD patient colonoids can be used to study the cytotoxic effects of the IBD-relevant cytokines IFN-γ and TNF-α on primary epithelium. We used a commercially available fluorescent cell death dye (SYTOX Green Nucleic Acid Stain), which can only enter cells that have a compromised cell membrane where it is then activated by binding to nucleic acids. We co-treated colonoids with cytokines and the fluorescent cell death dye and performed live cell imaging at 8 h and 24 h with an inverted epifluorescence microscope. Representative transmission/fluorescent overlay images at 8 h indicate that only the IFN-γ + TNF-α-treated colonoids are positive for fluorescent signal; however, there are only a small number of fluorescent cells (Figure 2A). Cell blebbing, a morphological indicator of cell death20, can also be observed in the IFN-γ + TNF-α condition. At 24 h, colonoids treated with IFN-γ + TNF-α display large regions positive for fluorescent signal (Figure 2A). There is also a clear breakdown in the morphology of the colonoid-the central lumen is no longer visible, and the epithelial barrier has been completely disrupted.
To quantify the cell death dye signal, we used open-source image analysis software to calculate the fluorescent intensity of each colonoid. We then normalized the data by expressing the mean of each condition as a percentage of the Max Toxicity treatment. At 8 h, homeostatic or background cell death in the BSA control colonoids was relatively low (7.7% of Max Toxicity) (Figure 2B). There were no statistically significant changes in cell death levels at this time point; however, conditions treated with TNF-α displayed a small increase in cytotoxicity (Figure 2B). After 24 h, cell death levels had increased for all cytokine-treated conditions. However, there was minimal change in cell death for the BSA Control condition between time points (7.5% of Max Toxicity at 24 h). The colonoids treated with IFN-γ + TNF-α had the largest increase in cell death levels compared to BSA control (29.4% of Max Toxicity). The difference in cell death levels between the combined treatment and the single cytokine treatments (IFN-γ, TNF-α) was highly significant. These results suggest the possibility of a cytotoxic synergistic interaction between IFN-γ and TNF-α at 24 h.
We used the CPI to quantify the cytotoxic interactions between cytokine treatments and to determine if they were synergistic. Interactions between cytokines are considered synergistic when the CPI value is <1, additive when =1, or antagonistic when >1. We calculated CPI values per time point (Figure 2C). At 8 h, the CPI value indicated slight synergism (0.99), with the CPI value decreasing substantially at 24 h (0.83). This analysis confirmed that the interaction between IFN-γ and TNF-α at 24 h was synergistic. Further, it illustrates how in this context synergism between IFN-γ and TNF-α is time-dependent.
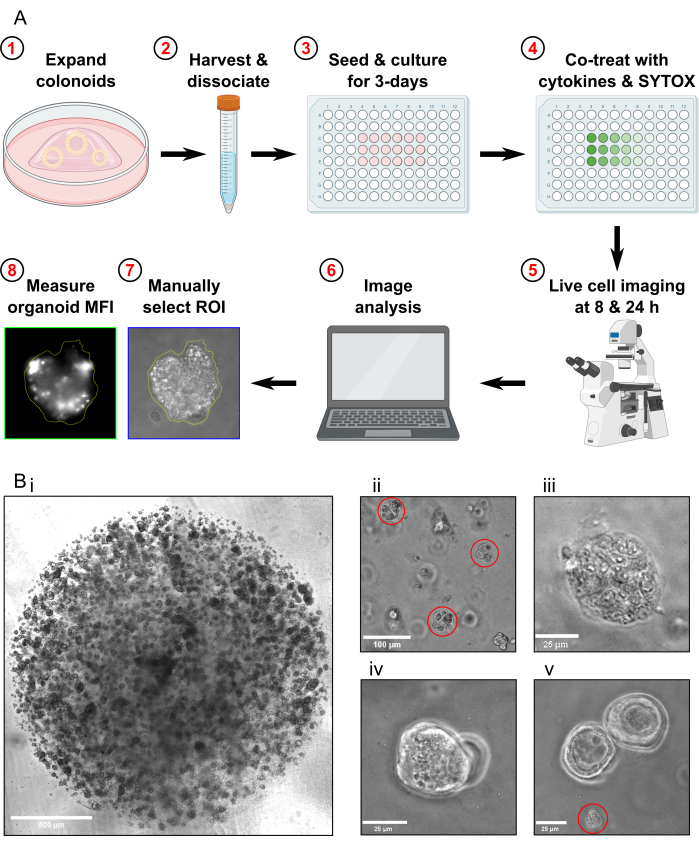
Figure 1: Schematic of experimental workflow and troubleshooting. (A) Schematic overview of protocol. (B) Representative images. (Bi) Light microscopy image illustrating the optimal density of the culture and optimal size of colonoids prior to passaging for an assay. Scale bar = 500 µm. (Bii) Light microscopy image illustrating optimal size of colonoid fragments after dissociation; fragments highlighted in red. Scale bar = 100 µm. (Biii) Light microscopy image of necrotic colonoid morphology after MT treatment (with Triton X-100). Scale bar = 25 µm. (Biv) Light microscopy image of two colonoids overlapping in the same focal plane. Scale bar = 25 µm. (Bv) Light microscopy image of colonoid cell debris present after passaging; debris highlighted in red. Scale bar = 25 µm. Abbreviations: ROI = region of interest; MFI = mean fluorescence intensity; MT = Max Toxicity. Please click here to view a larger version of this figure.

Figure 2: Quantitative analysis of cytokine-induced cell death in human CD colonoids. (A) Representative live microscopy images of CD colonoids treated with SYTOX Green Nucleic Acid Stain (fluorescent cell death dye) and cytokines at 8 h and 24 h; transmission and GFP (green color) channels overlayed. Colonoids were treated as follows: 1) PBS/BSA, 2) 10 ng/mL IFN-γ, 3) 10 ng/mL TNF-α, 4) 10 ng/mL IFN-γ + 10 ng/mL TNF-α. Scale bars = 25 µm. (B) Quantitative analysis of CD colonoids treated with the fluorescent cell death dye and cytokines at 8 and 24 h; data are expressed as a % of the MT condition. N = 2 CD colonoid lines, 11-16 colonoids imaged per condition. (C) CPI calculated per time point using the dataset from B, N = 2 CD colonoid lines. Data are expressed as means ± SE. In B, two-way ANOVA analysis was performed followed by Bonferroni post-tests, *P < 0.05, ***P < 0.001 as indicated. Abbreviations: CD = Crohn's disease; GFP = green fluorescent protein; CPI = coefficient of perturbagen interaction; PBS = phosphate-buffered saline; BSA = bovine serum albumin; TNF-α = tumor necrosis factor-alfa; IFN-γ = interferon-gamma; MT = Max Toxicity. Please click here to view a larger version of this figure.
Table 1: Composition of culture media for protocol. To prepare Organoid Proliferation Media, combine L-WRN conditioned media and Serum-free Media 1:1, then add supplements. Organoid Proliferation Media should be used within 2 weeks of preparation. Please note all complete media should be stored at 4 °C. Please click here to download this Table.
Table 2: Experimental 96-well plate layout and cytokine treatments. Please click here to download this Table.
