The general workflow to isolate MHC-peptide complexes for the analysis of immunopeptidomes by MS is illustrated in Figure 1. Representative results for the verification of the beads-binding efficiency of the antibody (Figure 2) (using the Y3 anti-H2Kb antibody) and the elution efficiency of the MHC-peptide complexes from the beads (Figure 3) (using the W6/32 anti-HLA-ABC antibody) are shown. Flow cytometry-based quantification assays21 were also applied to measure the absolute number of MHC class I molecules per EL4 cell (H2Kb and H2Db) and JY cell (HLA-ABC), as shown in Figure 4A.
The intra- and inter-individual reproducibility of the results using the current protocol are shown in Figure 4B–E. Representative results are shown for MHC class I peptides identified from 1 x 108 EL4 cells and 1 x 108 JY cells. Results were generated from two different lab members (User 1 and User 2). For User 1, the mean number of MHCI-specific peptides detected from EL4 and JY cells was 1341 and 6361, respectively; for User 2, 1933 and 7238, respectively (Figure 4B,D). The average coefficient of variation (CV) for the number of peptides detected across three different biological replicates/experiments varies from 1.9%-11% (Figure 4B,D). Although the CVs for the number of peptides detected across the three different experiments were relatively small, the identity of the peptides varied considerably (Figure 4C,E). Indeed, representative Venn diagrams show that the proportion of peptides that were reproducibly detected across three biological replicates ranged from 39% (User 2, EL4 cells) to 63% (User 1, JY cells) (Figure 4C,E and Supplementary Table 2).
Heatmaps generated by the MVP software show predicted MHC binding strength of the identified peptides using the NetMHCpan suite tools25,26,28. Two GibbsCluster routine options, which are termed 'Unsupervised GibbsCluster' and 'Allele-Specific GibbsCluster', are also performed by MVP to extract MHC peptide-binding motifs. Note that MVP has limitations; its primary goal is not to extract allele-specific motifs and annotate peptides in a highly accurate manner but to rather provide a bird's eye view on the overall quality, composition, and MHC-specificity of the samples.
For mouse MHC class I (H2Db and H2Kb) peptides in EL4 cells (Figure 5A; upper panels and Supplementary Data 1), a representative heatmap shows that 89% of all detected 8-12-mer peptides are predicted to be Strong Binders (SB: NetMHCpan %Rank <0.5) or Weak Binders (WB: NetMHCpan %Rank <2) for H2Db or H2Kb molecules. Sequence clusters generated from the 8-12-mer peptides are in concordance with reported logos for H2Db (asparagine at P5 and Leucine at P9) and H2Kb (phenylalanine at P5 and leucine at P8) (Figure 5)29. It is worth mentioning that a third dominant motif (histidine at P7 and leucine at P9) as well as additional 'artefactual' motifs can be observed using the M1 antibody (Supplementary Data 1). In fact, the M1 antibody is known to cross-react with the non-classical Qa2 molecule, and therefore, Qa2-associated peptides are also detected by MS (Supplementary Data 1). Here, for simplicity, Figure 5 focuses on showing the well-established peptide binding motifs for the two classical H2b alleles (i.e., H2Db or H2Kb) expressed in EL4 cells.
For human HLA class I (HLA-ABC) peptides in JY cells (Figure 5A; lower panels and Supplementary Data 2), a representative heatmap shows that 97% of all detected 8-12-mer peptides are predicted to be SB or WB for HLA-A*0201, -B*0702 or -C*0702. Peptides were clustered to visualize peptide binding motifs for HLA-A*0201 and -B*0702. Binding motif for HLA-C*0702 is not shown in Figure 5A because the C*0702 allele has a relatively low expression level. Therefore too few C*0702 peptides were isolated and identified to generate a representative C*0702 motif.Note that the C*0702 motif can be visualized in other studies30,31,32 or from the NetMHCpan 4.1 Motif Viewer website33.
For human HLA class II (HLA-DR) peptides in JY cells (Figure 5B; upper panels and Supplementary Data S3), a representative heatmap shows that 70% of all detected 9-22-mer peptides are predicted to be SB or WB for HLA-DRB1*0404 and -DRB1*1301. Peptide binding motifs for these two alleles are shown (Figure 5B). Note that the peptide-binding motifs shown here may not be in complete concordance with the recently reported logos for HLA-DRB1*0404 and -DRB1*130134. This discrepancy highlights the current inability of MVP/NetMHCpan to precisely annotate peptides to HLA class II alleles that are less characterized, such as HLA-DRB1*0404 and -DRB1*1301 expressed in JY cells. Additional information on class II peptide binding motifs can be found in other studies34,35 and from the NetMHCIIpan 4.0 Motif Viewer website36.
Finally, for mouse MHC class II (H2-IAd and H2-IEd) peptides in A20 cells (Figure 5B; lower panels and Supplementary Data 4), a representative heatmap shows that 87% of all detected 9-22-mer peptides are predicted to be SB or WB for H2-IAd or H2-IEd. Peptide binding motifs for these two alleles are in concordance with reported logos37.
Complete HTML reports generated by the MVP software to assess the overall quality and MHC-specificity of the samples are available in Supplementary Data 1–4.
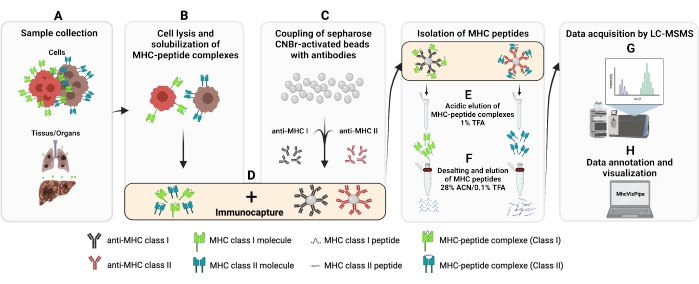
Figure 1: Schematic of the complete procedure for the isolation of MHC class I and II peptides. (A–B)100 million cells are pelleted and lysed with 0.5% Chaps buffer. (C) The cell lysate is centrifuged, and the supernatant is added to CNBr-sepharose beads coupled to the desired antibody beforehand and (D) incubated 14-18 h at 4 °C. (E) Following immunocapture, the beads are transferred into a polypropylene column and washed, and the MHC-peptide complexes are eluted with a 1% TFA solution. (F) The peptides are desalted and eluted using a C18 column. (G) Subsequently, peptides are speed vac dried and analyzed by tandem mass spectrometry. (H) The quality of the isolated MHC class I and II peptides can be assessed based on the HLA subtypes using the freely available MhcVizPipe software. Figure created with BioRender.com (NT22ZL8QSL). Please click here to view a larger version of this figure.
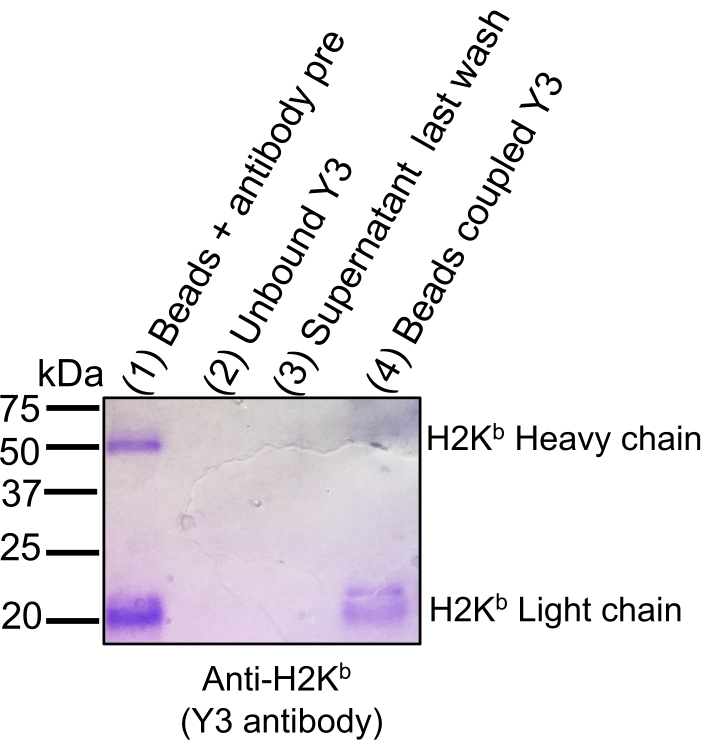
Figure 2: Coomassie gel staining to track antibody binding efficiency to sepharose CNBr-activated beads. Aliquots of equivalent volumes were loaded onto 12% SDS-PAGE gel followed by Coomassie blue staining: beads + antibody pre-coupling (1), unbound antibody following coupling step (2), supernatant following last wash after coupling (3), and beads coupled with antibody (4). The efficiency of binding is illustrated by a significant decrease in signal staining intensity of the light and heavy chains of H2Kb when beads are covalently bound (Lane 4) to the CNBr beads compared to the antibody before coupling (Lane 1). The figure has been reprinted and adapted from bioRxiv38. Please click here to view a larger version of this figure.

Figure 3: Western blotting for tracking MHC-peptide complexes following acidic elution from antibody-coupled CNBr-activated beads. Aliquots taken from the steps indicated in the protocol (1/100 of the total measured volume) were loaded onto a 12% SDS-PAGE gel and transferred onto nitrocellulose membrane: Beads + lysate following immunocapture and last wash (A); supernatant of last wash (B) and MHC-peptide complexes eluted from the beads (C). The strong detection signal of the MHC-peptide complexes in (C) using anti-HLA-ABC heavy chain antibody (Abcam, #ab 70328, 1:5000) confirmed the isolation of MHC-peptide complexes following acidic elution. Note that it is possible to assess the proportion of MHC-peptide complexes that were not captured by the antibody coupled beads by collecting the flow-through from step 3.1.6. An aliquot can be added to the western blot (not shown on this gel). Please click here to view a larger version of this figure.
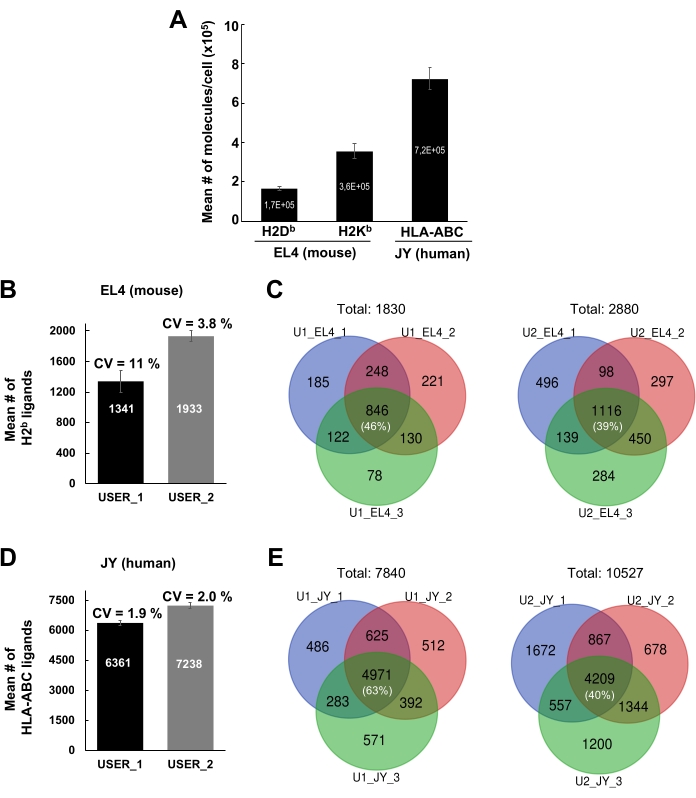
Figure 4: Identification of MHC class I peptides from JY and EL4 cells. (A) Histogram showing the absolute number of cell surface H2Db and H2Kb molecules per EL4 cell and of HLA-ABC molecules per JY cell. Quantification was performed by flow cytometry. Mean and standard error of the mean were obtained from three biological replicates. (B, D) Histogram showing the mean number and standard deviation of the mean of MHC class I peptides identified by two independent users (USER_1 [U1] and USER_2 [U2]). Mean number and standard deviation of the mean of MHC class I peptides detected from mouse EL4 cells (B) and human JY cells (D) are shown. Coefficients of variation (CV) across three independent biological replicates are indicated. (C, E) Venn diagrams showing the number of peptides that were reproducibly detected in EL4 (C) and JY cells (E) by two independent users (U1 and U2) across three independent biological replicates. Figure 4A has been reprinted and adapted from bioRxiv38. Please click here to view a larger version of this figure.
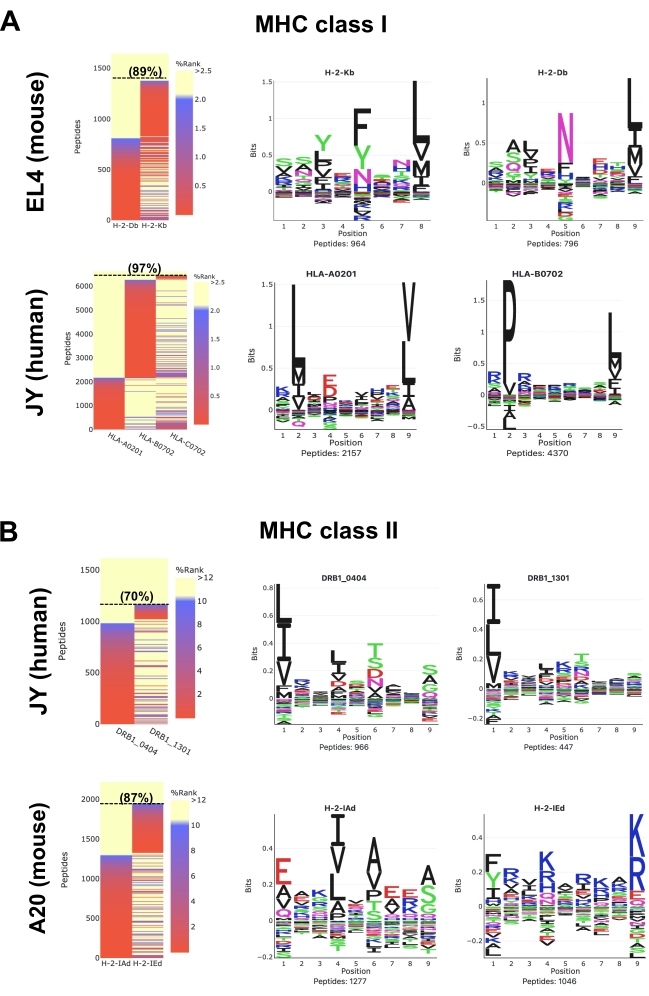
Figure 5: Visualization of immunopeptidomics data using the MVP software tool. Data analysis of mouse and human (A) MHC class I and (B) MHC class II peptides. Representative binding affinity heatmaps (left panels) and peptide binding motifs (right panels) are shown. Heatmaps colors represent the MHC binding affinity predicted by NetMHCpan 4.1 (%rank). Strong Binders are red (%rank <0.5), Weak Binders are blue (%rank <2) and Non-Binders are yellow (%Rank >2). Proportion (%) of 8-12mer peptides (A) and 9-22mer peptides (B) that are SB or WB is indicated in parenthesis. Peptide binding motifs were generated by MVP using the 'Allele-specific Gibbscluster' option. These representative results were obtained from 1 x 108 cells and by using the following antibodies and cell lines: M1 antibody and EL4 cells for mouse MHC class I peptides; W6/32 antibody and JY cells for human MHC class I peptides; M5 antibody and A20 cells for mouse MHC class II peptides; L243 antibody and JY cells for human MHC class II peptides. Refer to the Supplementary Data 1–4 to access the full HTML reports generated by the MVP software. Please click here to view a larger version of this figure.
Supplementary Data 1-4: Please click here to download this File.
Supplementary Data 1: HTML report generated by the MVP software from peptides that were isolated from 1 x 108 EL4 cells using the M1 antibody. Three biological replicates are shown. This report is related to Figure 4 and Figure 5 and representative peptides are listed in Supplementary Table 2.
Supplementary Data 2: HTML report generated by the MVP software from peptides that were isolated from 1 x 108 JY cells using the W6/32 antibody. Three biological replates are shown. This report is related to Figure 4, and Figure 5, and representative peptides are listed in Supplementary Table 2.
Supplementary Data 3: HTML report generated by the MVP software from peptides that were isolated from 1 x 108 JY cells using the L243 antibody. This report is related to Figure 5, and representative peptides are listed in Supplementary Table 2.
Supplementary Data 4: HTML report generated by the MVP software from peptides that were isolated from 1 x 108 A20 cells using the M5 antibody. This report is related to Figure 5, and representative peptides are listed in Supplementary Table 2.
Supplementary Table 1: List of buffers. Recipes for all the buffers used in the protocol are described. Please click here to download this Table.
Supplementary Table 2: Representative lists of peptides associated with H2Db/Kb, HLA-ABC, HLA-DR, and H2-IAd/IEd. This table contains the lists of representative MHC class I and II peptides isolated from mouse EL4 and A20 cell lines, respectively, and HLA class I and II from human JY cell line. These data have been deposited on the ProteomeXchange (PXD028633). Please click here to download this Table.
DATA AVAILABILITY:
Datasets used in this manuscript were deposited on the ProteomeXchange (http://proteomecentral.proteomexchange.org/cgi/GetDataset): PXD028633.
