Review and adhere to national and international ethical considerations and conditions related to the use of human tissues or cells before planning and executing any research activity involving this protocol.
NOTE: All steps of this protocol must be carried out in aseptic conditions. Biosafety Level 2 practices are the minimum requirement for the cultivation of RhEs. All necessary safety precautions must be taken when handling the chemicals/reagents described in this protocol.
1. Preparation of cell culture media
NOTE: There are three different types of serum-free culture media used for the cultivation of RhEs (Table 1): (i) the basal medium with low calcium level (60 µM Ca2+) used for the 2D culture of NHEKs; (ii) the submerged medium with high calcium level (1.5 mM Ca2+) used for the seeding of NHEKs into the cell culture insert system; and (iii) the air-liquid interface (ALI) medium with high calcium level (1.5 mM Ca2+), ascorbic acid, and keratinocyte growth factor (KGF).
| Medium | Medium information | Required quantity |
| Basal medium | Keratinocyte growth medium | 36 mL/24 wells |
| + 1 % [v/v] HKGS | ||
| + 1 % [v/v] 100x antibiotic-antimycotic | ||
| Submerged medium | Keratinocyte growth medium | 36 mL/24 wells |
| + 1 % [v/v] HKGS | ||
| + 1 % [v/v] 100x antibiotic-antimycotic | ||
| + 1.5 mM Ca2+ | ||
| Air-liquid interface medium | Keratinocyte growth medium | 216 mL/24 wells |
| + 1 % [v/v] HKGS | ||
| + 1 % [v/v] 100x antibiotic-antimycotic | ||
| + 1.5 mM Ca2+ | ||
| + 50 µg/mL ascorbic acid | ||
| + 10 ng/mL keratinocyte growth factor |
Table 1. Summary table of the different culture media used to cultivate RhEs. List of different culture media with supplements.
- Prepare the basal medium.
- Supplement the bottle of 500 mL of keratinocyte growth medium (Table of Materials) with 5 mL of human keratinocyte growth supplements (HKGS) in order to reach final concentrations of 0.2% [v/v] bovine pituitary extract (BPE), 0.2 ng/mL human recombinant epidermal growth factor (EGF), 0.18 µg/mL hydrocortisone, 5 µg/mL bovine transferrin, and 0.01 µg/mL of recombinant human insulin-like growth factor-I (IGF-I).
- Add 5 mL of 100x antibiotic-antimycotic solution containing 10,000 units/mL of penicillin, 10,000 µg/mL of streptomycin, and 25 µg/mL of amphotericin B.
- Prepare the submerged medium.
- Supplement the bottle of 500 mL of keratinocyte growth medium (Table of Materials) with 5 mL of HKGS in order to reach final concentrations of 0.2% [v/v] BPE, 0.2 ng/mL human recombinant EGF, 0.18 µg/mL hydrocortisone, 5 µg/mL bovine transferrin, and 0.01 µg/mL of human recombinant IGF-I.
- Add 5 mL of 100x antibiotic-antimycotic solution.
- Add 5 mL of a 0.144 M CaCl2 (calcium chloride) stock solution to reach a final concentration of 1.5 mM Ca2+.
NOTE: The calcium concentration is already increased during the submerged phase to stimulate the differentiation of the keratinocytes and initiate the stratification process49.
- Prepare the air-liquid interface (ALI) medium.
- Supplement one bottle of 500 mL of keratinocyte growth medium (Table of Materials) with 5 mL of HKGS in order to reach final concentrations of 0.2% [v/v] BPE, 0.2 ng/mL human recombinant EGF, 0.18 µg/mL hydrocortisone, 5 µg/mL bovine transferrin, and 0.01 µg/mL of human recombinant IGF-I.
- Add 5 mL of 100x antibiotic-antimycotic solution.
- Add 5 mL of a 0.144 M CaCl2 stock solution to reach a final concentration of 1.5 mM Ca2+.
- Add 1 mL of a 25 mg/mL ascorbic acid stock solution to reach a final concentration of 50 µg/mL ascorbic acid.
- Add 50 µL of a 100 µg/mL KGF in 1% [w/v] bovine serum albumin in phosphate-buffered saline (PBS) stock solution to reach a final concentration of 10 ng/mL KGF.
NOTE: Since ascorbic acid is sensitive to oxidation, it is recommended to use a stable ascorbic acid-derivative, such as magnesium l-ascorbyl-2-phosphate59 or L-ascorbic acid 2-phosphate sesquimagnesium60. If ascorbic acid is used, it is recommended to freshly supplement the ALI medium with ascorbic acid before each refresh.
2. Culture of NHEKs
NOTE: Since primary human keratinocytes remain proliferative upon their fourth or fifth passage61, NHEKs in their third passage are used for the cultivation of RhEs. Primary keratinocytes should be handled very carefully due to their high sensitivity. Careful and slow pipetting of cell suspensions at any time is very important, to not disturb the condition of the cells.
- Thaw a vial with 1 x 106 cryopreserved NHEKs in a water bath at 37 °C, by submerging part of the vial in the water. Incubate the vial for 1-2 minutes in the water bath, until only a small sliver of ice is visible.
CAUTION: Do not submerge the whole vial in the water bath to avoid contaminations. Do not thaw the cells longer than 2 minutes; this can reduce the cell viability. Wipe the vial with a 70% ethanol solution before transferring the tube into the laminar hood. - Resuspend the cells very carefully, by pipetting up and down 2-3 times. Transfer the cell suspension into two T75 flasks containing a total of 15 mL of pre-warmed thawing medium, resulting in a seeding density of 6.7 x 104 cells/cm2.
NOTE: For the first two passages and the thawing of cryopreserved NHEKs, cell culture medium is used according to the supplier's recommendations. - Place the flasks into the cell culture incubator (37 °C, 5% CO2, and 95% relative humidity (RH)).
- After approximately 24 hours, replace the thawing medium by the basal medium to remove dimethyl sulfoxide (DMSO) from the keratinocyte freezing solution.
- Refresh the basal medium every two days.
- After 4-6 days of cultivation, the cells should be around 80% confluent and ready for seeding in inserts for the cultivation of RhEs.
NOTE: Keratinocytes should be grown to maximum 80% confluence to preserve their proliferative capacity62. The number of cells to be thawed must take into consideration several parameters, such as the cell passage number, the cell viability upon thawing, the seeding efficiency as well as the doubling time.
3. Seeding of NHEKs
NOTE: This protocol is designed for use within a 24-well carrier plate format. If other plate formats are required (e.g., 12-well or 6-well format), optimizations in the seeding density and medium volume should be considered. Figure 1 summarizes a proposed timeline for RhE cultivation and shows the cultivation conditions.
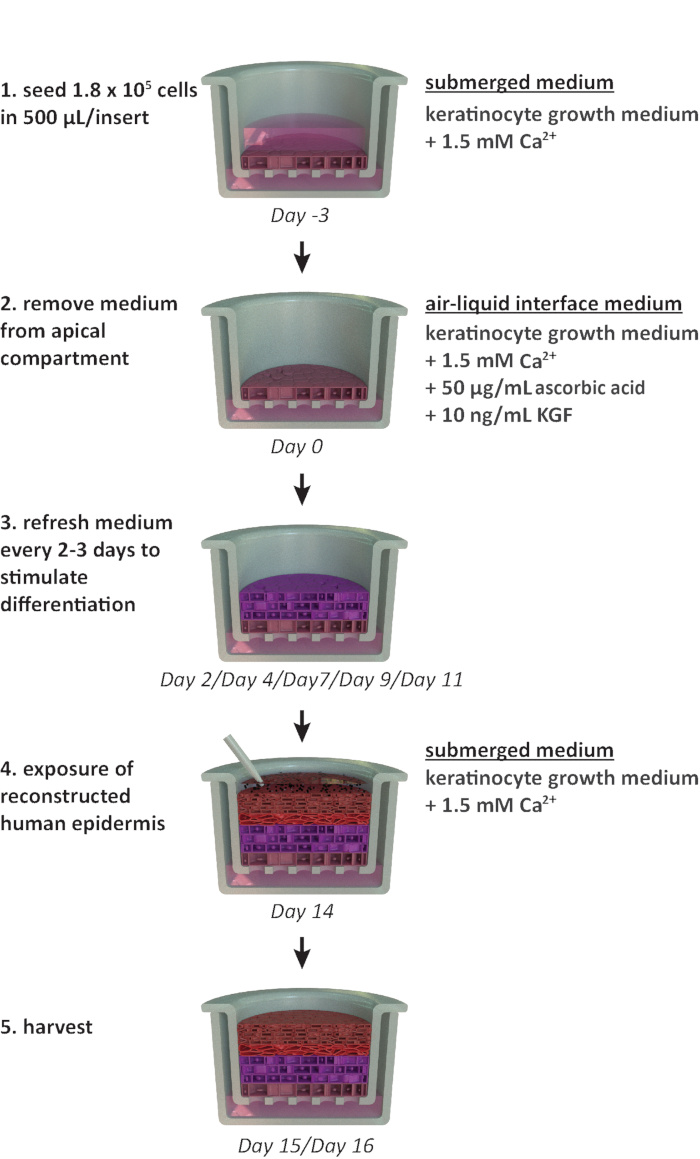
Figure 1: Schematic timeline of the reconstitution protocol. Presentation of the RhE model preparation, cultivation process, and application (exposure to chemical substance). The scheme includes the appropriate cell culture media types for each step. Please click here to view a larger version of this figure.
- Pre-fill 24-well plates with 1.5 mL of submerged medium, ideally using a dispenser pipette.
- Remove the basal medium from the T75 flasks used for the culture of the NHEKs.
- Rinse the cells by adding 5 mL of pre-warmed PBS to each T75 flask.
- Remove PBS from the flasks.
NOTE: This step is crucial, since the medium contains proteins and calcium that will inhibit the trypsin activity. - Add 2-3 mL of pre-warmed 0.05% [v/v] trypsin/ethylene diamine tetra acetic acid (EDTA) to each T75 flask. Make sure that the trypsin solution is equally distributed on the cell culture area of the flask.
CAUTION: The 2 mL volume is based on the 80% confluence mentioned above. Use 3 mL for flasks with a higher confluency. - Place the flasks for 4 minutes in the cell culture incubator (37 °C, 5% CO2, and 95% RH). Check whether the cells detach using the microscope at a 10x magnification. Rap the flask against the palm of the hand to help the cells release from the surface of the flask. Detached cells can be observed as rounded cells floating in the trypsin solution.
CAUTION: Do not incubate the cells in trypsin for longer than 6 minutes. Over-trypsinization can damage the cells and decrease their adherence63. - Once all the cells are detached, add an equal volume (i.e., 2-3 mL) of pre-warmed trypsin inhibitor to each T75 flask.
- Transfer the cell suspension from the flasks to a centrifuge tube.
- Rinse the flasks with 5 mL of pre-warmed PBS and transfer it to the centrifuge tube containing the cell suspension.
NOTE: Make sure that most of the cells are collected by checking the number of residual cells in the flasks under the microscope. The surface of the flask should be 95% empty. If this is not the case it is possible to repeat the trypsinization step (3.3-3.9). Note however that re-trypsinization should be avoided. - Centrifuge the harvested cells at 400 x g for 5 min.
- Carefully discard most of the supernatant, leaving approximately 100-200 µL in the tube.
CAUTION: Do not aspirate the pellet during this procedure. - Gently resuspend the pellet of cells in a low volume of submerged medium, pipette up and down 5-10 times to ensure a uniform cell suspension. Start with a low volume (i.e., 500 µL) to avoid the formation of cell aggregates and add up to 1 mL of submerged medium in total per initial T75 flask.
NOTE: Gently flick the tube with fingers to carefully dissolve a part of the cell pellet in the supernatant. - Count the cells in the suspension using the trypan blue exclusion method.
- Dilute 0.4% [v/v] trypan blue stain and the cell suspension in a 1:1 ratio, by adding 10 µL of 0.4% [v/v] trypan blue stain to 10 µL of cell suspension. Add 10 µL of the solution to a counting slide. Measure the cell count immediately after mixing the cell suspension with trypan blue, since trypan blue starts to decrease cell viability after exposure longer than 1 min65.
CAUTION: Trypan blue was shown to be a potential mutagen, carcinogen, and teratogen64. Handle the dye with care and dispose of the waste safely according to local laboratory regulations.
NOTE: An alternative approach to the use of trypan blue is the non-hazardous dye Erythrosin B66.
- Dilute 0.4% [v/v] trypan blue stain and the cell suspension in a 1:1 ratio, by adding 10 µL of 0.4% [v/v] trypan blue stain to 10 µL of cell suspension. Add 10 µL of the solution to a counting slide. Measure the cell count immediately after mixing the cell suspension with trypan blue, since trypan blue starts to decrease cell viability after exposure longer than 1 min65.
- Dilute the cell suspension with additional submerged medium to reach a concentration of 3.525 x 105 cells/mL in submerged medium by adding the volume V2 as shown in equation 1:
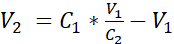
C1 = counted cell concentration in the cell suspension obtained in 3.12 (cells/mL)
V1 = volume used to resuspend the pellet of cells in 3.12 (mL)
C2 = targeted cell concentration in the suspension (i.e., 3.525 x 105 cells/mL)
V2 = volume to be added to reach the targeted cell concentration (mL)
NOTE: The surface area of the recommended culture insert is 0.47 cm2; therefore, the corresponding seeding density is 3.75 x 105 cells/cm2. - Perform a second cell count (C3) of the diluted solution obtained in step 3.14. Use equation 2 to calculate the cell suspension volume (V4) to be seeded into the culture insert:
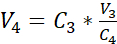
C3 = targeted cell concentration in the suspension (i.e., 3.525 x 105 cells/mL)
V3 = targeted volume of the cell suspension to be seeded in the culture insert (i.e., 0.5 mL)
C4 = counted cell concentration in the diluted suspension obtained in 3.14 (cells/mL)
V4 = actual volume of the cell suspension to be seeded in the culture insert (mL) - Hang the 24 cell culture inserts in the highest position of the recommended carrier plate and transfer the carrier plate to the 24-well plate pre-filled with submerged medium (cf. 3.1).
CAUTION: When transferring the carrier plate to the multi-well plate, ensure that no air bubbles are trapped between the insert membrane and the submerged medium from the basal compartment, as this will affect the feeding of the cells and ultimately compromise the RhE viability and morphology. - Add the determined volume V4 (from equation 2) of the cell suspension to each insert.
NOTE: It is recommended to use the reverse pipetting technique to accurately dispense the cell suspension to the culture inserts.
CAUTION: Make sure not to damage the membrane when dispensing the cell suspension into the culture insert. A precaution is to dispense the cell suspension along the wall of the insert system without touching the surface of the membrane. - After seeding, incubate the 24-well plates for 10-15 min at room temperature, to overcome an edge effect (i.e., non-uniform temperature distribution between all wells67). Do not move the plates during this time.
- Transfer the plates to the cell culture incubator (37 °C, 5% CO2, and 95% RH). The cells are maintained in submerged conditions for three days.
NOTE: To avoid tissue variability, do not stack the plates in the incubator after seeding to make sure that each insert receives the same amount of heat. After three days (i.e., during ALI cultivation), stacking of plates is possible.
4. Cultivation at Air-Liquid Interface
- After a three-day incubation in the cell culture incubator (37 °C, 5% CO2, and 95% RH), expose the cells that have adhered to the membrane surface to the ALI by removing the submerged medium from the apical compartment preferably using an aspiration system and a glass Pasteur pipette.
NOTE: Alternatively, the submerged medium from the apical compartment can be removed with a manual micropipette. - Fill new 24-well plates with 1.5 mL of fresh pre-warmed ALI medium and transfer the carrier plates with the culture inserts to the new multi-well plates.
- Transfer the multi-well plates back to the cell culture incubator (37 °C, 5% CO2, and 95% RH).
- Refresh the ALI medium every 2-3 days for 14 days.
- Perform the refresh in two steps: 1) prepare a new plate containing 1.5 mL/well of fresh pre-warmed ALI medium and 2) transfer the carrier plate to the new plate.
CAUTION: During the entire reconstitution procedure it is best not to remove the lid covering the carrier plate to keep the RhEs protected from potential contamination.
NOTE: The ALI step is crucial for the development of a stratified epidermal model as it allows terminal differentiation of the keratinocytes68. After going to ALI, a visual control of the inserts is required, to check whether there are 'leaky tissues': medium droplets on the tissue surface coming from the basolateral compartment. If the leakage happens at ALI day 3, gently remove the medium from the culture insert without touching the surface of the tissue. If the leakage persists, it is recommended to discard leaking tissues as it is an indication that there is no correct barrier formation in the RhE model. - At the end of the reconstitution process, the tissues can be exposed to various stressors to induce for example oxidative stress or inflammation. In parallel, they can be treated with chemical compounds or cosmetic ingredients. NOTE: During the exposure/treatment, tissues are usually maintained in submerged medium starting from ALI D14. When tissues are expected to be exposed/treated for a long period of time (i.e., 48-72 hours), it is recommended (i) to start the exposure/treatment earlier in the ALI cultivation process, such as D7-D9, to avoid the thinning of the viable layers and the thickening of the SC, and (ii) to incubate the tissues in ALI medium, to keep stimulating cell proliferation.
- For RhE harvesting, collect the tissues and cell culture medium at the timepoint of interest for histological analysis, viability assays, protein/RNA extraction, and enzyme-linked immunosorbent assays (ELISAs).
NHEKs cultured in 2D display a traditional morphology with a consistent polygonal shape (Figure 2A). As described above, NHEKs are seeded into culture inserts after reaching a confluency of approximately 80%. The morphology of the RhEs was analyzed using H&E staining and TEM. After 15 days at ALI, a fully stratified tissue is obtained as indicated by its four main epidermal layers: the SB, the SS, the SG, and the SC (Figure 2B). In the SB layer, the cells have a columnar shape. From the second layer on towards the upper layers of the RhE, NHEKs differentiate as observed by the changes in the cell morphology (from a columnar shape in the SB layer, towards a spinous shape in the SS layer). In the SG layer, the cells have a more flattened shape and display keratohyalin granules (KG) that are represented as purple dots in the cytoplasm. Their characteristic round and stellar shape is highlighted by white arrows on the H&E image (Figure 2C). The cells in the SC, are terminally differentiated and are completely flattened and lack a cell nucleus. The stratified RhEs have an overall thickness of 84.3 ± 2.4 µm and their SC has a thickness of 19.6 ± 3.2 µm (Figure 2D). These values are comparable to those reported for native human skin, i.e., 60-120 µm and 10-20 µm, respectively69. The number of viable layers is 6-7, which is lower compared to that of native human skin, being approximately 7-1470. Ultrastructural analysis of RhEs at different time points in the reconstitution protocol (i.e., 7, 10, 13, and 15 days) reveals the cornification process of the RhEs with an increased number of corneocyte layers over time (Figure 2E). After 15 days at the ALI, the SC of the RhE tissue is made of approximately 15-25 layers, which is comparable to the value reported for native human skin (i.e., 15-20 layers)69.
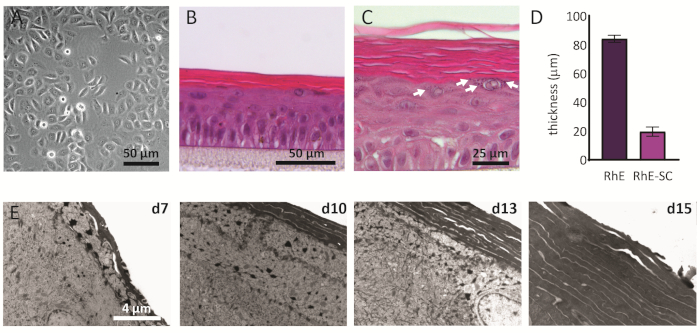
Figure 2: Primary keratinocytes and reconstructed human epidermis. (A) Phase-contrast microscopy image of primary keratinocytes before seeding onto inserts. Scale bar is 50 µm. (B-C) H&E bright field microscopy image of RhE. Scale bar is 50 µm (B) and 25 µm (C). (D) Quantification of thickness of RhE and SC (mean ± SEM, n=3). (E) Transmission electron microscopy images of RhE cross-sections after 7, 10, 13, and 15 days at ALI. Scale bar is 4 µm. Please click here to view a larger version of this figure.
According to their differentiation stage, NHEKs growing in 3D show different protein expression profiles according to their differentiation stage. The expression of proteins specific for early-stage keratinocyte differentiation (i.e., keratin 10), late-stage keratinocyte differentiation (i.e., involucrin, loricrin, and filaggrin), and keratinocyte adhesion (i.e., desmoglein 1) in RhEs was determined using IF staining. Involucrin expression appears more predominantly located in the SG layer since its expression is initiated earlier during the differentiation process (Figure 3D), whereas filaggrin and loricrin are expressed in the upper layers (Figure 3B-C). Keratin 10 expression was found in all the viable layers, except of the SB layer (Figure 3E). RhEs display functional desmosomal junctions, as indicated by the expression of desmoglein 1 in the intercellular space of the viable epidermal layers (Figure 3F). To conclude, all five markers are expressed and located in the appropriate epidermal layers and translate to a healthy epidermal differentiation process.
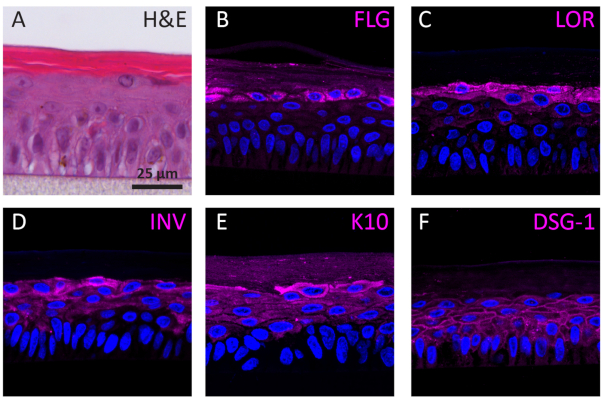
Figure 3: Epidermal differentiation, tissue adhesion, and tissue integrity of reconstructed human epidermis. (A) H&E bright field microscopy image of RhE. Confocal fluorescence microscopy images of (B) filaggrin (FLG), (C) loricrin (LOR), (D) involucrin (INV), (E) keratin 10 (K10), and (F) desmoglein 1 (DSG-1) represented in magenta. Nuclei staining (DAPI) is represented in blue. Scale bar is 25 µm. Please click here to view a larger version of this figure.
The barrier properties of the RhE model was investigated by assessing both the tissue viability and integrity. The tissue integrity was determined after 15 days by measuring the TEER using a voltohmmeter. The 2567 ± 415 Ω.cm2 values recorded for the RhEs translate the formation of a continuous barrier (Figure 4A). Those values are in range with those reported for RhE models71,72,73,74. Additionally, the required exposure time for a cytotoxic reference chemical (i.e., Triton X-100) to reduce the tissue viability by 50% (ET50) was determined with a thiazolyl blue tetrazolium bromide (MTT) assay. The ET50 value measured for the RhE was 2.1 hours. This value falls within the acceptance range of other 3D epidermal models that are qualified for reliable prediction of irritation classification (OECD Guideline 439)19.
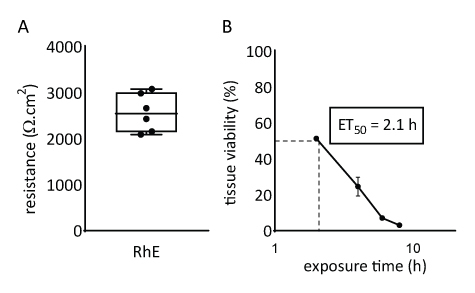
Figure 4: Barrier properties of reconstructed human epidermis. (A) Tissue integrity measured with transepithelial electrical resistance (mean ± SEM, n=6). (B) ET50 determined by measuring tissue viability (i.e., MTT assay) upon topical exposure to 78.3 µL of 1% Triton X-100 (mean ± SEM, n=3).
Responsiveness of RhEs was investigated upon known proinflammatory stimuli. RhEs were treated systemically, i.e., addition of stimuli in the medium of the basolateral compartment, using 100 µg/mL Escherichia coli lipopolysaccharide (LPS) and 40 ng/mL tumor necrosis factor alpha (TNF-α). After 24 hours of stimuli, the cell culture medium was collected. The cytotoxicity was measured using a lactate dehydrogenase (LDH) assay and compared to values of a known membrane disruptor, the Triton X-100 detergent (Figure 5). A significant increase (p < 0.05, one-way ANOVA, Dunnett's multiple comparison test) was shown in LDH activity in RhEs treated with Triton X-100. LPS and TNF-α treatments both showed not be cytotoxic.
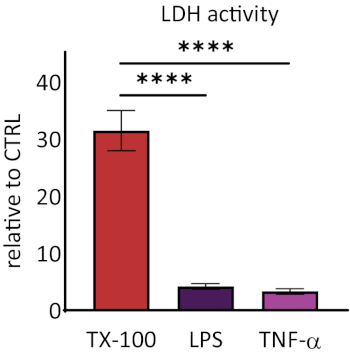
Figure 5: The cytotoxicity measured via lactate dehydrogenase (LDH) assay. Data are presented as relative values to control, untreated tissues (CTRL); mean ± SEM, n=9 (Triton X-100), n=8 (LPS), n=3 (TNF-α). Significance was tested with one-way ANOVA, Dunnett's multiple comparison test. Asterisk denotes statistically significant difference compared to CTRL, ****p < 0.0001). Please click here to view a larger version of this figure.
The release of interleukin 1 alpha (IL-1α) and interleukin 8 (IL-8) in the RhE medium was quantified using ELISAs. Figure 6 shows both the quantified and relative IL-1α and IL-8 release by the RhEs upon challenge with LPS and TNF-α. LPS treatment resulted in a statistically significant (p < 0.05, unpaired Student's T-test) induced release of IL-8 (9.6 ± 1.0 fold increase) and IL-1α (2.7 ± 1.3 fold increase). TNF-α did not significantly induce IL-8 release, even though a tendency of increased IL-8 levels was observed (2.3 ± 0.8 fold increase). However, TNF-α did significantly (p < 0.05, unpaired Student's T-test) trigger IL-1α release (1.8 ± 0.5 fold increase).
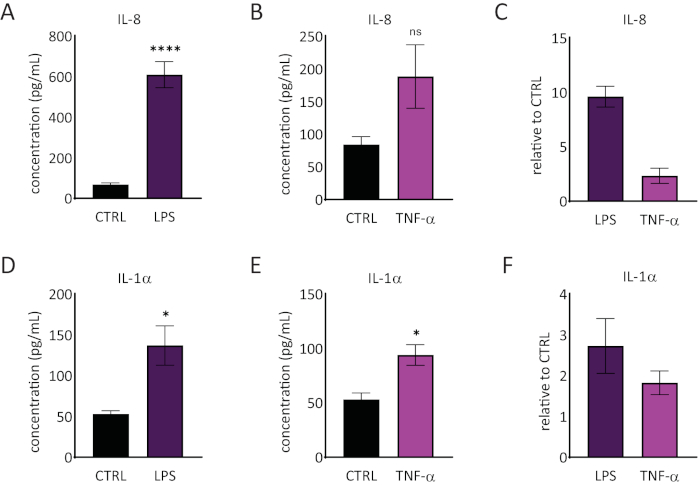
Figure 6: Proinflammatory responses in the reconstructed human epidermis. Concentrations of IL-8 release by the RhE upon a 24-hour challenge with LPS (A) and TNF-α (B). Data is represented as mean ± SEM, n=8 (LPS), n=3 (TNF-α). (C) Data is represented as the mean of relative value compared to control, untreated tissues (CTRL) ± SEM, n=8 (LPS), n=3 (TNF-α). The concentration of IL-1α release of the RhE upon a 24-hour challenge with LPS (D) and TNF-α (E). Data is represented as mean ± SEM, n=8 (LPS), n=3 (TNF-α). (F) Data is represented as the mean of relative value compared to CTRL ± SEM, n=4 (LPS), n=3 (TNF-α). Significance was tested by an unpaired Student's T-test. Asterisk denotes statistically significant differences compared to CTRL, *p < 0.05, ****p < 0.0001). Please click here to view a larger version of this figure.
