Transduction and Expansion of Primary T Cells in Nine Days with Maintenance of Central Memory Phenotype
Summary
We outline a 9-day protocol for the transduction and expansion of rhesus macaque peripheral blood mononuclear cells which yields cells with excellent co-expression of the genes of interest in sufficient number for infusion studies of cell efficacy.
Abstract
Emerging immunotherapies to treat infectious diseases and cancers often involve transduction of cellular populations with genes encoding disease-targeting proteins. For example, chimeric antigen receptor (CAR)-T cells to treat cancers and viral infections involve the transduction of T cells with synthetic genes encoding CAR molecules. The CAR molecules make the T cells specifically recognize and kill cancer or virally infected cells. Cells can also be co-transduced with other genes of interest. For example, cells can be co-transduced with genes encoding proteins that target cells to specific locations. Here, we present a protocol to transduce primary peripheral blood mononuclear cells (PBMCs) with genes encoding a virus-specific CAR and the B cell follicle homing molecule chemokine receptor type 5 (CXCR5). This procedure takes nine days and results in transduced T cell populations that maintain a central memory phenotype. Maintenance of a central memory or less differentiated phenotype has been shown to associate with persistence of cells post-infusion. Furthermore, cells produced with this method show high levels of viability, high levels of co-expression of the two transduced genes, and large enough quantities of cells for immunotherapeutic infusion. This nine-day protocol may be broadly used for CAR-T cell and other T cell immunotherapy approaches. The methods described here are based on studies presented in our previous publications.
Introduction
Cellular immunotherapies are emerging as a new means to treat diseases including cancers and infectious diseases. These immunotherapy methods often involve genetically manipulating therapeutic cells to express specific molecules. For example, chimeric antigen receptor (CAR) T cells are engineered to express a CAR molecule that has an extracellular domain that specifically binds molecules on diseased cells and a signaling domain that triggers the immune cells to kill the diseased cell. CAR T cells are being used successfully in cancer immunotherapies, and have been particularly effective in treating B cell leukemias1,2,3. CAR-T cells are also being developed to treat viral infections such as HIV. HIV-specific CARs target the envelope proteins on the surface of virally infected cells4. Immunotherapeutic cells can also be engineered to express homing molecules to target therapeutic cells to specific tissue locations. We have developed vectors that transduce both a virus-specific CAR as well as the lymphoid follicle homing molecule CXCR55.
Viral replication of some viruses, including human immunodeficiency virus (HIV) and simian immunodeficiency virus (SIV), is concentrated within B cell follicles in secondary lymphoid tissues6. B cell follicles are somewhat immune privileged sites in which low levels of virus-specific CTL permit ongoing viral replication7,8. For these reasons, targeting HIV/SIV-specific T cells into the follicle through expression of CXCR5 is a strategy for elimination of the follicular reservoir of virally infected cells9,10. Specifically, we are targeting SIV-specific CAR T cells to B cell follicles via CXCR5 co-expression.
In this protocol we describe a method to transduce CAR/CXCR5 and expand PBMCs to produce CAR/CXCR5 T cells of sufficient quantities for therapeutic infusion. Cells transduced with these methods maintain a central memory phenotype. Studies have shown that cells with a less differentiated phenotype, such as central memory T cells and T memory stem cells better persist than more differentiated cells11,12. In addition, many protocols designed to produce adoptively transferred cells have relatively long culture times that result in cells that have a more differentiated phenotype and reduced persistence13. Furthermore, adoptively transferred cells with long culture times accumulated within the lungs instead of target lymphoid tissues in rhesus macaque14,15. In the methods we describe and demonstrate here, we rapidly transduce and expand rhesus macaque PBMCs to produce transduced T cells that maintain a central memory phenotype.
Both CD4 and CD8 T cells are included in our immunotherapy approach. This mixed cell population was chosen because, in clinical trials, the absence of CD4+ T cells in the CD8+ T cell product was correlated with a failure of infused CD8 T cells to persist 16. The methods described here begin by activating PBMCs with anti-CD3 and anti-CD28 antibodies which potently stimulate the T cell receptor and provide a co-stimulatory signal to avoid clonal anergy17,18.
After activation, cells are transduced with a gammaretroviral vector encoding a virus-specific CAR and the lymphoid homing molecule CXCR5. Transduced cells are then expanded using plates designed for non-adherent cell propagation which have a gas permeable membrane at the base. This membrane permits gas exchange at the bottom of the well leading to enhanced cell survival and nutrient availability19. Using these methods, sufficient functional cells can be produced in a 9-day timeframe for in vivo infusion studies. While this protocol is designed for transducing and expanding rhesus macaque T cells, with slight modification of the species-specific activating antibodies, it can be used with cells from other species. These methods are based on our previously published studies9,20.
Protocol
NOTE: This rhesus PBMC transduction protocol requires the use of a gammaretroviral vector. Note that gammaretrovirus can either be produced in the lab or outsourced to a viral production company. In lab production can be carried out with the protocol outlined in step 1 and the virus can be titered as outlined in step 2. Whether the virus is produced on site or by a production company, an MOI (ratio of infectious virions to cells in culture) should be determined, as outlined in step 3, for each newly produced viral preparation and the cells of interest.
CAUTION: Work with gammaretroviral vectors or other retroviral vectors requires proper safety precautions such as use of lab coats and double layers of gloves. All handling of the virus must occur in the biosafety hood. All pipets should be decontaminated in a solution of 10% bleach for 30 min. All liquid infectious waste should be decontaminated with a final concentration of 10% bleach. Secondary containment, with a seal that prevents release of microbiological aerosols, is required for centrifugation
1. Producing the Gammaretroviral Stock
- Plate 293T cells for transfection. A full virus preparation requires 12 T75 flasks at 4.5 x 106 cells/flask in Dulbecco's modified Eagle medium (DMEM) + 10% fetal bovine serum (FBS) without antibiotics.
- Allow the cells to grow overnight at 37 °C before transfection.
- Dilute 60 μL of transfection reagent in 1.5 mL reduced serum media. Prepare one tube for each flask. Incubate for 5 minutes at RT.
- For each flask, prepare a tube containing 1.5 mL of reduced serum media and the following plasmids: 9.0 μg of a transfer plasmid containing the gene(s) of interest, 3 μg of RD114 and 1.2 μg of VSV-G (envelope plasmids), and 3 µg of gag/pol.
NOTE: The envelope and gag/pol genes are needed for retroviral packaging. For our retroviral production, we add both the RD114 envelope and, for added stability, a small amount of VSV-G envelope. - Add diluted plasmids (step 1.4) to diluted transfection reagent (step 1.3). Mix gently. Incubate for 20 min at room temperature.
- Feed cells 5 mL of fresh complete media and add 3 mL of transfection reagent/plasmid mixture dropwise per flask and incubate for 5–6 h at 37 °C.
- Add an additional 5 mL of fresh medium to the flasks and incubate for 42–43 h at 37 °C.
- After a total transfection time of 48 h, collect media and centrifuge at 1282 x g for 3 min at 4 °C to remove cell debris.
- Aliquot into appropriate volumes for future use, flash freeze the virus in an ethanol/dry ice bath and store at -80 °C.
2. Titering the Gammaretrovirus
NOTE: Several dilutions of virus may be needed to obtain a valid titer.
- Plate 293T cells at 600,000 cells/well in a 6-well plate and incubate the plates for 24 h at 37 °C.
- Remove the medium from the cells and add desired amount of DMEM + 10% FBS to the 293T (2 mL total volume of virus).
- Add the virus to the corresponding well and swirl gently to mix. For example, add 200 µL, 100 µL, 50 µL and 25 µL respectively to 4 wells. Include a well without virus for flow cytometry (mock sample).
- Incubate for 48 h at 37 °C.
- Trypsinize the 293T cells by adding 0.5 mL of Trypsin-EDTA and incubating for 4 min at 37 °C. Stop the trypsin by adding 1.5 mL of DMEM + 10% FBS. Count the cells and determine viability with an automated cell counter or a hemocytometer.
- To use the cell counter, add 10 µL of cells to 10 µL of trypan blue, mix, load the chamber slide, and insert into the counter. Push the "capture" button to count the cells.
- Collect 0.5–1 x 106 cells and evaluate levels of CAR and CXCR5 by flow cytometry (see step 9.5). Expression of CD4 or MBL along with CXCR5 will identify transduced 293T cells co-expressing the CAR and CXCR5. See reactive antibodies in step 9.5.1.
- Calculate viral titer.
NOTE: Choose the sample with a transduction level of 20% or less for calculation of titer.- Use the following formula to calculate titer:
transduction units/mL = (number of cells in culture)(% of cells transduced)/volume of virus added to the culture
- Use the following formula to calculate titer:
3. Determining the Optimal MOI for a Newly Produced Viral Preparation and the Cells of Interest
- Follow the transduction protocol (sections 4–9) with primary PBMCs and twofold dilutions of the gammaretroviral preparation.
- Assess the level of expression of the genes of interest by flow cytometry (step 9.5).
- Choose a concentration of virus that allows maximal transduction without loss of cell viability.
4. Preparing Plates for T Cell Activation by Coating Wells with Anti-CD3 Antibodies
- Prepare anti-macaque CD3 antibody (FN18) by diluting the stock to 10 µg/mL in phosphate-buffered saline (PBS).
- Dispense 2 mL/well in 6-well plates. Incubate at 37 °C for 2 h. Alternatively, the plates can be incubated overnight at 4 °C.
- Aspirate the PBS and unbound antibodies. Rinse wells twice with 2 mL of PBS and use immediately to plate PBMCs.
5. Stimulating Rhesus PBMCs with Plate-bound Anti-CD3 and Soluble Anti-CD28
- Thaw primary rhesus PBMCs in a 37 °C waterbath, with gentle agitation, until just a small amount of ice remains.
- In the hood, gently pipette cells into a conical tube. Rinse the vial with 1 mL of basic medium and add it slowly to the cells. Next, slowly add an additional 9 mL of warm basic medium to the cells.
NOTE: This step can be scaled up but never thaw more than 4 vials at one time. - Centrifuge at 600 x g for 5 min at 22 °C to pellet the cells. Aspirate the supernatant and then resuspend the pellet in a small amount of growth medium. Choose a volume such that the concentration of cells will exceed 2 x 106 cells/mL at this point since the final concentration should be 2 x 106 cells/mL.
- Stain cells with trypan blue and count the cells to determine the number of viable cells.
NOTE: This can be done with a standard hemocytometer or an automated cell counter. We use an automated cell counter which displays the viability and number of live cells.- To use the counter, add 10 µL of cells to 10 µL trypan blue, mix, load the chamber slide and insert into the counter. Push the "capture" button to count the cells.
- Calculate the total number of cells by multiplying cell concentration by volume and then dilute the cells to 2 x 106 cells/mL in growth medium. Prior to plating, add anti-CD28 to a final concentration of 5 µg/mL in order to provide a necessary co-stimulatory signal for T cell activation.
- Pipette cells into anti-CD3 coated plates. Add 3–6 x 106 cells per well (a good target is 4 x 106 cells in 2 mL media per well) and incubate for 2 days at 37 °C in 5% CO2.
6. Preparing Fibronectin-coated Plates
NOTE: PBMC and T cells express integrin receptors VLA-4 or VLA-5 and are good targets for fibronectin-mediated transduction.
- Prior to coating the plates, first prepare fibronectin reagent by diluting it 1:100 in sterile PBS.
- Pipette 2 mL of the sterile fibronectin solution into each well of a 6-well plate. Gently rock plates for 2 h at room temperature.
NOTE: Non-treated, cell culture-grade tissue culture plates or dishes should be used in this step. - Remove the fibronectin solution by aspiration and then block with 1 mL of sterile 2% bovine serum albumin (BSA, Fraction V) in PBS. Rock the plates at room temperature for 30 min.
- Aspirate the BSA solution and wash the plate with 2 mL of PBS. After aspiration of the PBS, the fibronectin-coated plates can be sealed with seal wrap and stored at 4 °C for up to a week.
NOTE: We usually prepare the plates one day before transduction.
7. Transduction of activated rhesus PBMCs
CAUTION: Work with viral vectors or with rhesus macaque PBMCs requires utilization of proper safety precautions such as use of lab coats and double layers of gloves. All handling of the virus and the cells must occur in the biosafety hood. All pipets should be decontaminated in a solution of 10% bleach for 30 min. All liquid infectious waste should be decontaminated with a final concentration of 10% bleach. Secondary containment with a seal that prevents release of microbiological aerosols, is required for centrifugation.
- Attach the gammaretroviral transducing vector to the fibronectin-coated well.
- Warm a centrifuge to 32 °C by running at 2000 x g for about 30 min.
- Warm both serum free and growth media in a 37 °C waterbath.
- Thaw the virus on ice or by gently swirling the vial in a 37 °C waterbath until only small amount of ice remains. To avoid degradation of the viral preparation, do not allow the contents to warm up.
- Dilute the retrovirus to a predetermined optimal multiplicity of infection (MOI) in serum-free medium.
NOTE: With our gammaretrovirus and rhesus PBMC, we have found that an MOI of 0.5 is optimal.- Example dilution: To coat 4 wells with virus and add 1.5 x 106 cells, (4)(1.5 x 106)(0.5) = 3 x 106 TU are needed. If the viral titer is 1.1 TU/mL, then use 2.7 mL virus in 5.3 mL media for a total volume of 8 mL.
- Add 2 mL diluted retrovirus to each well of the fibronectin-coated plate.
- For negative control mock-transduced cells, add media alone to the fibronectin-coated wells.
- Place the plates in the pre-warmed 32 °C centrifuge in microplate carriers with biosafe covers and centrifuge at 2000 x g for 2 h.
NOTE: Virus coated plates can be used immediately or stored, with virus, at 4 °C overnight. - For immediate use, aspirate the virus preparation from the wells and add 2 mL of growth medium. Keep the virus-coated wells from drying.
- Plate the stimulated PBMCs.
- Check the cells using an inverted microscope. The cells should look healthy, meaning that they are round, bright and refract light. They should also show a clumped appearance indicating stimulation.
- Collect the target cells and transfer them to a 50 mL conical tube. Rinse the well once with 1 mL growth media and add to the conical tube. Count the number of living cells with the automated cell counter as in step 5.4.
NOTE: Stimulated cells will be clumped and this clumping may lead to difficulties in accurately counting the cells. - Pellet the cells in the tubes by centrifuging at 600 x g for 5 min at 32 °C. Aspirate the media from the cell pellet and resuspend the cells in growth medium at a concentration of 1.5 x 106 cells/mL.
- To each virus-coated well (prepared in the step 7.1) add 1 mL of cell suspension for a total of 1.5 x 106 cells/3 mL. Note that each well already contains 2 mL growth medium. Perform the same steps for mock-transduced cells but plate them onto fibronectin-coated wells which received no virus.
- Centrifuge the plates at 1000 x g for 10 min at 32 °C.
- Incubate the plates for 48 h at 37 °C under 5% CO2.
8. Expansion of the transduced cells
- Draw the contents of the wells up and down with a 5 mL pipet in order to resuspend the cells and then transfer to a 50 mL conical tube.
- Add 1 mL of growth media to each well and draw up and down with a sterile transfer pipet to remove adherent cells.
- Count cells to determine the total cell number and viability using an automated cell counter as in step 5.4.
- If desired, remove 1 x 106 cells for flow cytometry to assess gene expression and phenotype. See step 9.5.1 and the Table of Materials for recommended antibodies and gating strategy.
- Centrifuge the cells at 600 x g for 10 min at 25 °C.
- Aspirate the media and resuspend the cells in expansion media to a concentration of 1 x 106 cells/mL. Seed each gas-permeable well of a 6-well plate with 5 mL of cells. Carefully layer an additional 25 mL of expansion media per well.
NOTE: We add the cells in a small volume and then layer remaining media so that the cells remain on the bottom of the well. - Incubate the cells undisturbed at 37 °C in 5% CO2 incubator for 4 days.
9. Collection of expanded cells and evaluating the phenotype of the transduced cell population
- Collect cells from gas permeable wells by removing and discarding 20 mL media. Caution should be taken to avoid disturbing the cells.
NOTE: We expand the cells for only 4 days and collect the cells at this point. However, if additional days of culture are desired, remove 20 mL of media without disturbing the cells and replace it with 20 mL of fresh expansion media. - Pipet the remaining media up and down to dislodge the cells. The media should be very cloudy if cells have grown well.
- Using a sterile transfer pipet, rinse each well with 3 mL media to collect any residual cells.
- Count the cells with the automated counter or a hemocytometer to check for viability and cell number.
- Perform flow cytometry to determine transduced gene expression and cell phenotype.
- In this example, determine rhesus macaque PBMC phenotype using antibodies directed against CD4 (M-T477), a clone that is reactive with both endogenous rhCD4 and the rhCD4-MBL CAR; against CD3 (SP34-2) and CD8 (RPA-T8) which stain total T cells and CD8 T cells respectively; against the death receptor CD95 (DX2) and the co-stimulatory molecule CD28 (CD28.2) which are used to determine the memory phenotype of the cells; and against CXCR5 (MU5UBEE) and MBL (3E7) to detect the CXCR5 and CD4-MBL CAR on the transduced cells.
- Assess viability was assessed with a fixable viability dye.
- Prepare cells for flow cytometry.
- Make up an antibody master mix for the total number of tubes. Bring up to total volume using PBS.
- Add the cells to flow tubes. Place 0.5–1 x 106 cells per tube. Include negative controls such as unstained cells as well as stained mock transduced cells.
- Add 2 mL of PBS to the cells, centrifuge at 400 x g for 5 min at room temperature (RT) and decant.
- Add 100 µL of antibody mix to the tubes and incubate for 30 min at RT.
- Add 2 mL of PBS, centrifuge at 400 x g for 5 min at RT and decant.
- Add 300 µL of 1% paraformaldehyde and incubate for 15 min.
- Centrifuge at 400 x g for 5 min at RT and decant. Add 300 µL of PBS and mix.
- On a flow cytometer, acquire 150,000 events for each sample.
- Analyze the data with flow analysis software.
NOTE: The gating strategy was previously published20.- For identification of transduced cells, gate cells on lymphocytes, singlets, live, CD3+, and MBL+CXCR5+.
- For identification of the central memory population, gate cells on lymphocytes, singlets, live CD3+, CD8+, CD28+ CD95+.
- Use cells as needed. Cells can be used for in vitro assays of function, such as a transwell migration assay9 or in vitro cell killing assays5,9. Cells can be used for infusion into animals. Alternatively, freeze any remaining cells in 90% FBS with 10% dimethyl sulfoxide (DMSO) for later use or analysis
NOTE: While a target dose for infusion of adoptively transferred cells into a rhesus macaque is not yet established, published adoptive transfer studies in non-human primates have used 0.6 to 1.2 x 107 cells/kg21, 1 to 5 x 108 cells/kg22 and 1.4 to 8 x 108 cells/kg10. With this range as a guideline, enough cells for infusion can be produced with this 9-day protocol.
Representative Results
Cell production
Results presented in this publication are similar to those in our previously published work9,20. Utilizing the protocol presented here, we expect at least an 8-fold expansion of cells between days 5 and 9 (median 11.1 fold, range 8.7–13 fold). Gas permeable wells were seeded at a starting density of 5 x 106 cells and after 4 days of growth, we achieved a median density of 55.6 x 106 cells per well (range 43.5–64.8 x 106) (Figure 1A). Cell counting with Trypan Blue exclusion shows that the cells maintain high viability throughout the protocol (day 9 median 85.8%, range 83–95%) (Figure 1B). We find animal to animal variability in the ability of the cells to expand; however, with a starting population of 50–100 x 106 cells on day 1, we have used this protocol to produce enough cells for rhesus macaque infusion of 1–2 x 108 cells/kg.
Co-expression of the transduced genes was monitored with flow cytometry (Figure 2A). Expression of the CAR and CXCR5 on the surface of the transduced cells was relatively stable between days 5 and 9 of this expansion protocol. On day 5, a median of 42.8% (range 36.5–46.9%) of the cells were transduced with both CD4-MBL CAR and CXCR5 while on day 9, median expression was 47.6% (range 44.5–64.5%) with a single round of transduction (Figure 2B). Although some protocols call for a second round of transduction, we have not seen a benefit in terms of total transduced cells at the conclusion of the protocol (data not shown).
Cell phenotype
Transduced cells were analyzed by flow cytometry to monitor their memory phenotype. Prior to transduction and expansion, naive, central memory and effector memory populations are identified (data not shown) but the naïve population is lost with culturing and the cells are primarily central memory cells by day 5 (Figure 3A) and day 9 (Figure 3B) as identified by CD95+ CD28+ expression. The use of this rapid transduction and expansion protocol has produced cells which are primarily central memory cells and thus will be more likely to proliferate and persist when infused into a test animal.
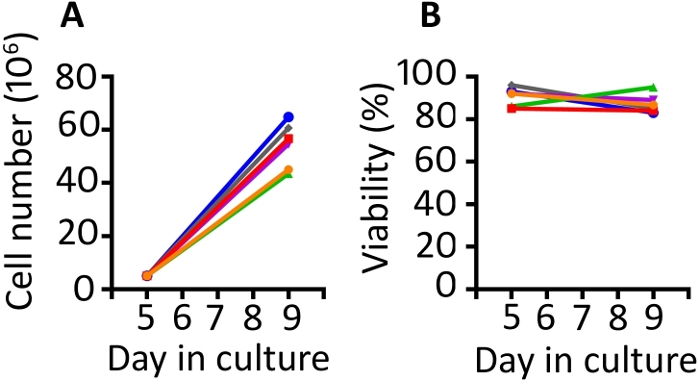
Figure 1: The transduction and expansion protocol produces abundant transduced cells which are highly viable. (A) Expansion of the transduced cells from 6 different animals from days 5 to 9 in the gas permeable vessels and (B) viability of the transduced PBMC from the same 6 animals. Trypan blue exclusion using an automated cell counter was used to monitor cell number and viability. Please click here to view a larger version of this figure.
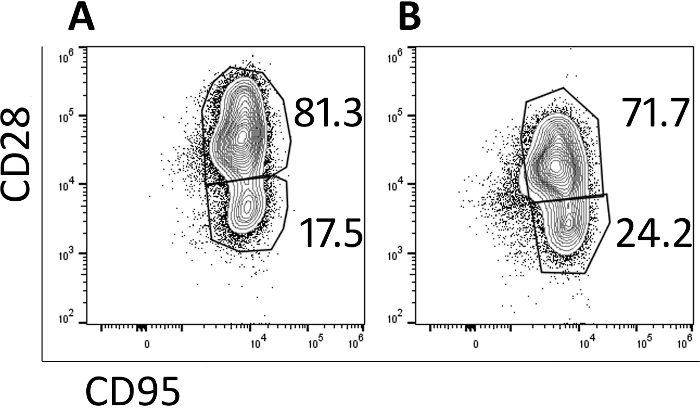
Figure 2: The transduction protocol produces cells which maintain co-expression of the two transduced genes. (A) A representative flow plot of rhesus PBMC on day 9 of the transduction and expansion protocol demonstrating expression of both the CD4-MBL CAR and CXCR5. (B) Expression of the CD4-MBL CAR and CXCR5 on transduced rhesus PBMC from 6 different animals on days 5 and 9 in culture as measured by flow cytometry. Gates were set on live, CD3+ cells. Transduced cells are identified as MBL+ CXCR5+. Please click here to view a larger version of this figure.
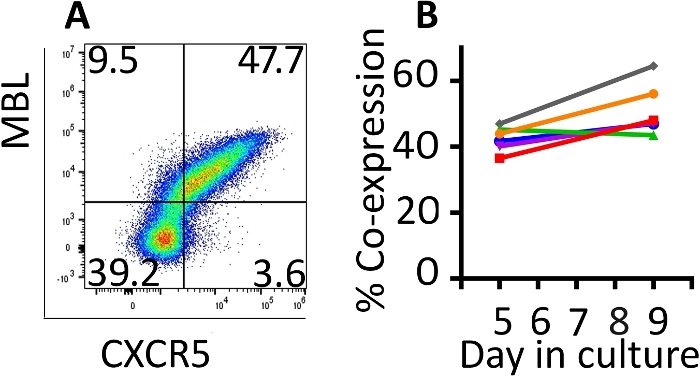
Figure 3: The majority of the cells produced in the 9-day protocol have a central memory phenotype. Representative flow cytometry plots of (A) day 5 and (B) day 9 CAR/CXCR5-transduced rhesus PBMC. Gates were set on live, CD3+, CD8+ cells. Central memory was defined as CD28+ CD95+. Effector memory was defined as CD28–, CD95+. Please click here to view a larger version of this figure.
Discussion
This protocol outlines a T cell immunotherapy production strategy which utilizes a gammaretroviral vector to transduce rhesus macaque PBMC leading to T cell population that expresses an antiviral CAR as well as the follicular homing receptor, CXCR5. With a total of 8 days of ex vivo culture time, viable, functional CAR T cells are produced in a quantity that is within the published range10,21,22 used for infusion into non-human primates for preclinical efficacy testing.
The success of the transduction protocol relies on both healthy, stimulated PBMCs and on quality preparations of gammaretrovirus. In order to achieve successful stimulation and transduction, it is essential that proper care is taken in freezing and transport of the PBMCs after collection. Ideally, if the cells are collected off site, they are shipped in liquid nitrogen and quickly placed in long term liquid nitrogen storage. PBMCs must be mitotically active in order to be successfully transduced by a gammaretrovirus. One should monitor visually for clustering of the cells after stimulation with anti-CD3/anti-CD28. A failure to activate properly will cause low efficiency of transduction. It is also important to monitor the viability in the stimulated cells. Poor viability after the stimulation step usually leads to a less successful transduction. Whether virus preparations are produced in the lab or outsourced, they should be stored at -80 °C in single use aliquots to avoid freeze thaw cycles that can damage the virus and impair transduction efficiency. The virus must be titered so that consistent amounts of virus can be used in each experiment. When using the virus for transduction, thaw the virus quickly and store on ice until needed. The choice of promoter, enhancer and envelope proteins may all impact transduction efficiency and expression of transgene in target cells. Thus, a MOI must be empirically determined. In addition, the use of a media designed to support T cells and plates with gas permeable wells for expansion have led to excellent expansion of the transduced cells. However, there is animal to animal variability in the cell expansion rate. We recommend a trial study to determine the ability of a particular cell preparation to be transduced and expanded. Expansion levels are consistent in both small-and large-scale transductions.
With the use of this protocol, we have produced up to 2.5 x 109 cells for infusion into test animals. Although we have found it unnecessary to scale-up the protocol at this time, the use of 6 well plates is a limitation to large-scale cell preparation and the protocol would require modification to produce greater numbers of cells. Examples of potential modifications include carrying out the transduction in a fibronectin-coated culture bag to increase the number of cells transduced and to utilize a larger gas permeable culture vessel for the expansion step. Although we have not yet made these alterations, they utilize commercially available products and are feasible modifications to the current protocol.
Using this method, we have produced transduced cells from PBMC isolated from uninfected animals, SIV-infected animals and from SIV-infected animals treated with antiretroviral therapy (ART). However, we have noted a reduction in transduction efficiency in the ART treated cells20. This reduction is presumably due to the inhibition of reverse transcriptase and/or integrase by the drugs. Transductions of cells from ART-treated animals will require modifications to this protocol such as reducing the intracellular levels of the ART drugs by stopping ART for several days prior to collecting the PBMC or through the use of an alternative vector which is not impacted by the ART drugs commonly in use.
It is important that cells produced for immunotherapy be of a minimally differentiated phenotype so that they will persist post-infusion12. Although many protocols for adoptive transfer of cells require long culture times, a reduced ex vivo culture time has been correlated with both a reduction in differentiation and an improvement in CAR T cell function13. The relatively rapid timeframe of this transduction and expansion protocol allows maintenance of the desired central memory phenotype while still producing sufficient cells for testing of their immunotherapeutic potential20.
The goal of the production strategy for this T cell immunotherapy product is to manufacture T cells that will recognize SIV-infected cells, will traffic to the site of viral replication in the B cell follicle and will persist in the animal resulting in a long-term functional cure without need for anti-retroviral drugs. For translation to a human immunotherapy product, this protocol can be modified to transduce human T cells through the use of human-specific antibodies and cytokines and the implementation of GMP standards with the ultimate goal of a producing a functional cure for HIV.
Divulgazioni
The authors have nothing to disclose.
Acknowledgements
This study was supported by NIH grants 5R01AI096966-06S1 (PS, EC, and EB), 1UM1AI26617 (PS, EC and EB), P51OD011106/P51RR000167 (ER), MN REACH grant 5U01HL127479-03 (PS), 1R01A143380-01 (PS and EB), 1UM14126617 (PS and EC) as well as funds provided by the NIAID Division of Intramural Research and the NIH Intramural AIDS Targeted Antiviral Program. Anti-CD3 and anti-CD28 used in these studies was provided by the NIH Nonhuman Primate Reagent Resource (R24 OD010976, U24 AI126683). IL-2 used in these studies was provided by The NCI Preclinical Repository. We thank our collaborators in this CD4-MBL CAR/CXCR5 project, Dr. Elizabeth Connick at the University of Arizona, Dr. Edward A Berger at NIAID, NIH, Dr. Eva G Rakasz at the Wisconsin National Primate Research Center and Dr. Geoff Hart and Ms. Preethi Haran at the University of Minnesota, Dr. Leslie Kean at Harvard Medical School and Dr. Catherine Bollard at the Children's Research Institute. We also thank Dr. Scott McIvor at the University of Minnesota, Dr. Christopher Peterson at the Fred Hutchinson Cancer Center, Dr. Matthew Trivett at the NIH, Dr. Agne Taraseviciute at Seattle Children's Hospital, and Dr. Conrad Russell Cruz at The Children's Research Institute for their very helpful assistance in optimizing this protocol. We also gratefully acknowledge Ms. Chi Phan and Ms. Jhomary Alegria-Berrocal at the University of Minnesota for gammaretroviral production, and Ms. Kim Weisgrau at the University of Wisconsin-Madison for isolation of rhesus macaque PBMC.
Materials
| Gammaretrovirus preparation | |||
| 0.025% Trypsin, 0.01% EDTA | Gibco | R-001-100 | |
| 293T cells | ATCC | CRL-3216 | |
| 6 well plates, treated | CytoOne | CC7682-7506 | |
| DMEM | Gibco | 10569-010 | |
| Heat-inactivated FBS | Hyclone | Sh30088.03 | |
| Lipofectamine | Invitrogen | 11668019 | transfection reagent |
| Opti-Mem | Invitrogen | 31985070 | reduced serum media |
| pBS-CMV-gagpol | Addgene | 35614 | A gift from Dr. Patrick Salmon |
| pMSGV1 containing CAR P2A CXCR5 | custom order from GenScript | ||
| RD114 | A gift from Dr. Ed Berger | ||
| T75 flasks | CytoOne | CC7682-4875 | |
| VSV-G | pMD.G | A gift from Dr. Scott McIvor | |
| T cell stimulation | |||
| 6 well plates, untreated | CytoOne | CC7672-7506 | |
| Anti-CD28 | NHP Reagent Resource | Clone: CD28.2 | |
| Anti-macaque CD3 | NHP Reagent Resource | Clone: FN18 | |
| Phosphate buffered saline | Gibco | 14190-144 | |
| Rhesus macaque PBMC or CD8 T cells | WNPRC | Primary cells | |
| For Fibronectin coating | |||
| 6 well plates, untreated | CytoOne | CC7672-7506 | |
| BSA (Fraction V) | HyClone | SH 30574.02 | |
| RetroNectin (1 mg/ml) | TaKaRa | T100A | |
| For T cell Expansion | |||
| G-Rex 6 Well Plate | Wilson Wolf | P/N 80240M | Plates with gas permeable wells |
| Media Components | |||
| b mercaptoethanol | Gibco | 21985-023 | |
| Heat-inactivated FBS | Hyclone | Sh30088.03 | |
| IL-2 | NCI Preclinical Repository | ||
| Penicillin/Streptomycin/Glutamine | Gibco | 10378-016 | |
| X-Vivo-15 medium | Lonza | 04-418Q | |
| Variations of Media used | |||
| Basic medium: | X-Vivo 15 medium, 10% heat-inactivated FBS, 1 x Penicillin/Streptomycin/L-Glutamine | ||
| Expansion medium: | Growth medium + 50 mM b mercaptoethanol | ||
| Growth medium: | Basic medium + 50 IU/ml IL-2 | Completion of media by addition of anti-CD28, IL-2 or b-mercaptoethanol should occur on the day of use. | |
| Cell counting | |||
| Countess cell counting chambers | Invitrogen | AMQAF1000 | |
| Countess II FL Automated Cell Counter | Invitrogen | T10282 | |
| Trypan blue, 0.4% solution | Invitrogen | T10282 | |
| Flow Cytometry | |||
| Alexa Fluor 647 Antibody Labeling Kit | Invitrogen | A20186 | for conjugation of MBL antibody |
| anti-CD28-BV605 | BD Biosciences | 562976 | |
| anti-CD3-AF700 | BD Biosciences | 557917 | |
| anti-CD4-FITC | BD Biosciences | 556615 | |
| anti-CD8-BV788 | BD Biosciences | 563824 | |
| anti-CD95-PerCP Cy5.5 | BD Biosciences | 561655 | |
| anti-CXCR5-PE | eBioscience | 12-1985-42 | |
| anti-MBL | Invitrogen | MA1-40145-S6 | |
| Flow Analysis software | FlowJo, LLC | FlowJo v10 | |
| Flow Cytometer | Beckman | CytoFlex | |
| Live/Dead Near IR | Invitrogen | L10119 | |
| Other equipment | |||
| Aerosolve canisters to contain aerosol leakage | Beckman | SX4750 | Safety equipment |
| Beckman Allegra Centrifuge | Beckman | Sterilgard e3 | |
| Cell culture incubator | Thermo Fisher | Everlast 247 | |
| Class II Laminar flow hood | Baker | Heracell Vios 160i | |
| Extra-Safe Disposable lab coat | Fisher Scientific | 359232 | Personal protective equipment |
| Microplate carriers with biocertified covers | Beckman | SX4750A | Safety equipment |
| Rocking platform | Benchmark | C10228 | |
| Swinging bucket rotor | Beckman | X13-R | |
| X-Gen Nitrile gloves | Genesee | Personal protective equipment |
Riferimenti
- Klebanoff, C. A., Rosenberg, S. A., Restifo, N. P. Prospects for gene-engineered T cell immunotherapy for solid cancers. Nature Medicine. 22 (1), 26-36 (2016).
- Lim, W. A., June, C. H. The Principles of Engineering Immune Cells to Treat Cancer. Cell. 168 (4), 724-740 (2017).
- Sadelain, M., Rivière, I., Riddell, S. Therapeutic T cell engineering. Nature. 545 (7655), 423-431 (2017).
- Kuhlmann, A., Peterson, C. W. Chimeric antigen receptor T-cell approaches to HIV cure. Current Opinion in HIV and AIDS. 13 (5), 446-453 (2018).
- Ghanem, M. H., Bolivar-Wagers, S., et al. Bispecific chimeric antigen receptors targeting the CD4 binding site and high-mannose Glycans of gp120 optimized for anti-human immunodeficiency virus potency and breadth with minimal immunogenicity. Cytotherapy. 20, 407-419 (2018).
- Folkvord, J. M., Armon, C., Connick, E. Lymphoid Follicles Are Sites of Heightened Human Immunodeficiency Virus Type 1 (HIV-1) Replication and Reduced Antiretroviral Effector Mechanisms. AIDS Research and Human Retroviruses. , (2005).
- Connick, E., Mattila, T., et al. CTL Fail to Accumulate at Sites of HIV-1 Replication in Lymphoid Tissue. The Journal of Immunology. 178 (11), 6975-6983 (2007).
- Connick, E., Folkvord, J. M., et al. Compartmentalization of Simian Immunodeficiency Virus Replication within Secondary Lymphoid Tissues of Rhesus Macaques Is Linked to Disease Stage and Inversely Related to Localization of Virus-Specific CTL. The Journal of Immunology. 193 (11), 5613-5625 (2014).
- Haran, K. P., Hajduczki, A., et al. Simian immunodeficiency virus (SIV)-specific chimeric antigen receptor-T cells engineered to target B cell follicles and suppress SIV replication. Frontiers in Immunology. 9 (MAR), 1-12 (2018).
- Ayala, V. I., Deleage, C., et al. CXCR5-Dependent Entry of CD8 T Cells into Rhesus Macaque B-Cell Follicles Achieved through T-Cell Engineering. Journal of Virology. 91 (11), e02507-e02516 (2017).
- Redeker, A., Arens, R. Improving adoptive T cell therapy: The particular role of T cell costimulation, cytokines, and post-transfer vaccination. Frontiers in Immunology. 7 (SEP), 1-17 (2016).
- Klebanoff, C. A., Gattinoni, L., Restifo, N. P. Sorting Through Subsets. Journal of Immunotherapy. 35 (9), 651-660 (2012).
- Ghassemi, S., Nunez-Cruz, S., et al. Reducing Ex Vivo Culture Improves the Antileukemic Activity of Chimeric Antigen Receptor (CAR) T Cells. Cancer Immunology Research. 6 (9), 1100-1109 (2018).
- Minang, J. T., Trivett, M. T., Bolton, D. L., Trubey, C. M., Estes, J. D., Li, Y., et al. Efficacy of Adoptively Transferred Central and Effector Memory-Derived Autologous Simian Immunodeficiency Virus-Specific CD8+ T Cell Clones in Rhesus Macaques during Acute Infection. The Journal of Immunology. 184 (1), 315-326 (2010).
- Bolton, D. L., Minang, J. T., et al. Trafficking, persistence, and activation state of adoptively transferred allogeneic and autologous Simian Immunodeficiency Virus-specific CD8(+) T cell clones during acute and chronic infection of rhesus macaques. Journal of immunology (Baltimore, Md. : 1950). 184 (1), 303-314 (2010).
- Patel, S., Jones, R. B., Nixon, D. F., Bollard, C. M. T-cell therapies for HIV : Preclinical successes and current clinical strategies. Cytotherapy. 18 (8), 931-942 (2019).
- Chambers, C. A., Allison, J. P. Costimulatory regulation of T cell function. Current Opinion in Cell Biology. 11 (2), 203-210 (1999).
- Schwartz, R. H. A cell culture model for T lymphocyte clonal anergy. Science. 248 (4961), 1349-1356 (1990).
- Bajgain, P., Mucharla, R., et al. Optimizing the production of suspension cells using the G-Rex M series. Molecular Therapy – Methods and Clinical Development. 1, 14015 (2014).
- Pampusch, M. S., Haran, K. P., et al. Rapid transduction and expansion of transduced T cells with maintenance of central memory populations. Molecular Therapy – Methods and Clinical Development. 16, 1-10 (2019).
- Taraseviciute, A., Tkachev, V., et al. Chimeric antigen receptor T cell-mediated neurotoxicity in nonhuman primates. Cancer Discovery. 8 (6), 750-763 (2018).
- Berger, C., Sommermeyer, D., et al. Safety of targeting ROR1 in primates with chimeric antigen receptor-modified T cells. Cancer Immunology Research. 3 (2), 206-216 (2015).
