Quaternary Structure Modeling Through Chemical Cross-Linking Mass Spectrometry: Extending TX-MS Jupyter Reports
Summary
Targeted cross-linking mass spectrometry creates quaternary protein structure models using mass spectrometry data acquired using up to three different acquisition protocols. When executed as a simplified workflow on the Cheetah-MS web server, the results are reported in a Jupyter Notebook. Here, we demonstrate the technical aspects of how the Jupyter Notebook can be extended for a more in-depth analysis.
Abstract
Protein-protein interactions can be challenging to study yet provide insights into how biological systems function. Targeted cross-linking mass spectrometry (TX-MS), a method combining quaternary protein structure modeling and chemical cross-linking mass spectrometry, creates high-accuracy structure models using data obtained from complex, unfractionated samples. This removes one of the major obstacles to protein complex structure analysis because the proteins of interest no longer need to be purified in large quantities. Cheetah-MS web server was developed to make the simplified version of the protocol more accessible to the community. Considering the tandem MS/MS data, Cheetah-MS generates a Jupyter Notebook, a graphical report summarizing the most important analysis results. Extending the Jupyter Notebook can yield more in-depth insights and better understand the model and the mass spectrometry data supporting it. The technical protocol presented here demonstrates some of the most common extensions and explains what information can be obtained. It contains blocks to help analyze tandem MS/MS acquisition data and the overall impact of the detected XLs on the reported quaternary models. The result of such analyses can be applied to structural models that are embedded in the notebook using NGLView.
Introduction
Protein-protein interactions underpin the structure and function of biological systems. Having access to quaternary structures of proteins can provide insights into how two or more proteins interact to form high-order structures. Unfortunately, obtaining quaternary structures remains challenging; this is reflected in the comparatively small number of Protein DataBank (PDB) entries1 containing more than one polypeptide. Protein-protein interactions can be studied with technologies such as X-ray crystallography, NMR, and cryo-EM, but obtaining a sufficient amount of purified protein under conditions where the methods can be applied can be time-consuming.
Chemical cross-linking mass spectrometry was developed to obtain experimental data on protein-protein interactions with fewer restrictions on sample preparation as mass spectrometry can be used to acquire data on arbitrarily complex samples2,3,4,5,6,7,8,9. However, the combinatorial nature of the data analysis and the relatively small number of cross-linked peptides require that the samples be fractionated before analysis. To address this shortcoming, we developed TX-MS, a method that combines computational modeling with chemical cross-linking mass spectrometry10. TX-MS can be used on arbitrarily complex samples and is significantly more sensitive compared to previous methods10. It accomplishes this by scoring all data associated with a given protein-protein interaction as a set instead of interpreting each MS spectrum independently. TX-MS also uses up to three different MS acquisition protocols: high-resolution MS1 (hrMS1), data-dependent acquisition (DDA), and data-independent acquisition (DIA), further providing opportunities to identify a cross-linked peptide by combining multiple observations. The TX-MS computational workflow is complex for several reasons. First, it relies on multiple MS analysis software programs11,12,13 to create protein structure models14,15. Second, the amount of data can be considerable. Third, the modeling step can consume significant amounts of computer processing power.
Consequently, TX-MS is best used as an automated, simplified computational workflow through Cheetah-MS web server16 that runs on large computational infrastructures such as computer clouds or clusters. To facilitate the interpretation of the results, we produced an interactive Jupyter Notebook17. Here, we demonstrate how the Jupyter Notebook report can be extended to yield a more in-depth analysis of a given result.
Protocol
1. Submit workflow at https://txms.org.
- Go to https://txms.org and click "Use Cheetah-MS."
- To submit workflow, you need to provide two PDB files and one MS/MS mzML or MGF file. You can also click on the "load sample data" to see the demo version of the workflow.
NOTE: Please look at the manual page of the webserver for detailed information on how to submit a job. The web server supports different non-cleavable cross-linker agents, up to 12 post-translational modifications (PTMs), options related to computational modeling and MS data analysis. Small help buttons are also designed on the submit page to show more information regarding each option.
2. Run Cheetah-MS.
NOTE: Convert the vendor-specific formats to mzML or MGF using the ProteoWizard MSConvert software19.
- Upload the MS data to https://txms.org. Then, click on "Choose file" and select the MS data, which must be in the mzML/MGF data formats18.
NOTE: Example data are available on https://txms.org. These data are also directly accessible through zenodo.org, DOI 10.5281/zenodo.3361621. - Upload two PDB files to https://txms.org. Click on "Choose file" and select the PDB files to upload.
NOTE: If no experimental structures exist, create models using, for example, SWISS-MODEL20 if homologue structures are available, or trRosetta21,22 or Robetta23,24 web servers for de novo structure predictions. - Submit a new workflow. Click on "Invia" to receive a job identifier tag. Then, follow the form to the results section using this tag.
NOTE: Computing the result takes time, so please wait until the workflow finishes, and store the job identifier tag to return to the results page. The computation is carried out on remote computational infrastructure. If you want to run TX-MS locally, please refer to Hauri et al.10. - Inspect the Jupyter Notebook report using the online viewer. Then, scroll down to "Report" in the results section using the job identifier tag.
3. Install JupyterHub.
- Install docker as instructed at https://docs.docker.com/install/.
- Download the JupyterHub docker container with the Jupyter openBIS25 extension. The general command is "docker pull malmstroem/jove:latest," but might differ on other platforms.
NOTE: For a general discussion on how to download containers, please refer to https://www.docker.com/get-started. It is also possible to download the container from zenodo.org, DOI 10.5281/zenodo.3361621.
NOTE: The Jupyter openBIS extension source code is available here: https://pypi.org/project/jupyter-openbis-extension/. - Start the docker container: docker run -p 8178:8000 malmstroem/jove:latest.
NOTE: The port that JupyterHub uses by default is 8000. This port is configurable, and the commands above need to be adjusted accordingly if changed. Port 8178 is an arbitrary choice and can be changed. The example URLs provided below need to be adjusted accordingly. - Go to the following address: http://127.0.0.1:8178. Log in using the username "user" and the password "user."
NOTE: The address http://127.0.0.1 implies that the docker container is running on the local computer. If the docker container is run on a server, use the server's IP address or URL (e.g., https://example.com). The docker container is based on Ubuntu Bionic 18.04, JupyterHub 0.9.6, and Jupyter openBIS extension 0.2. It is possible to install this in other operating systems, but this was not tested.
4. Download the report.
- Create a new notebook by clicking New| Python 3 using the menu located near the top right part of the page. This will open a new tab with a notebook called Untitled (or something similar).
- Click "Configure openBIS Connections" in the Jupyter tool menu.
- Fill in the name: txms; URL: https://txms.org; user: guest; password: guestpasswd.
- Click "Connect."
- Choose the new connection and click "Choose Connection."
- Search for the report template (e.g., /CHEETAH/WF70) and click Download.
NOTE: You need to adjust the report template based on the results and report you obtained from running your job on the Cheetah-MS web server. - Rerun the report by clicking Cell | Run All.
5. Extend the report.
- Add a new cell at the bottom: Cell | Insert Below.
- Type in the wanted code. For an example, please see the Representative Results section below.
- Execute the cell by pressing "Shift-Enter."
Representative Results
TX-MS provides structural outputs supported by MS-derived experimental constraints. It works by combining different MS data acquisition types with computational modeling. Therefore, it is helpful to parse each MS data separately and provide visualization of the output structure. Supplementary Data 1 contains an example notebook that can parse DDA and DIA data produced as TX-MS output. Users can select the XL of interest. By running the notebook, the MS2 spectrum of that XL will be shown where different colors help to discriminate between fragments related to the first peptide, second peptide, and the combinatorial fragment ions. The XL can also be mapped to the structure using the NGLView widget embedded in a Jupyter Notebook.
Another cell in this notebook can help users to parse and visualize DIA data. However, visualizing DIA data is more difficult because the analyzed data need to be prepared in the correct format.
Figure 1 shows an example structure of M1 and albumin with top XLs mapped on the structure. TX-MS obtained all XLs after parsing hrMS1, DDA, and DIA data, and the RosettaDock protocol provided the computational models.
As this report is a Jupyter Notebook, any valid Python code can be added to new notebook cells. For example, the code below will create a histogram over the MS2 counts, indicating how well supported each cross-link is by the underlying data.
import seaborn as sns
sns.distplot(ms2['count']);
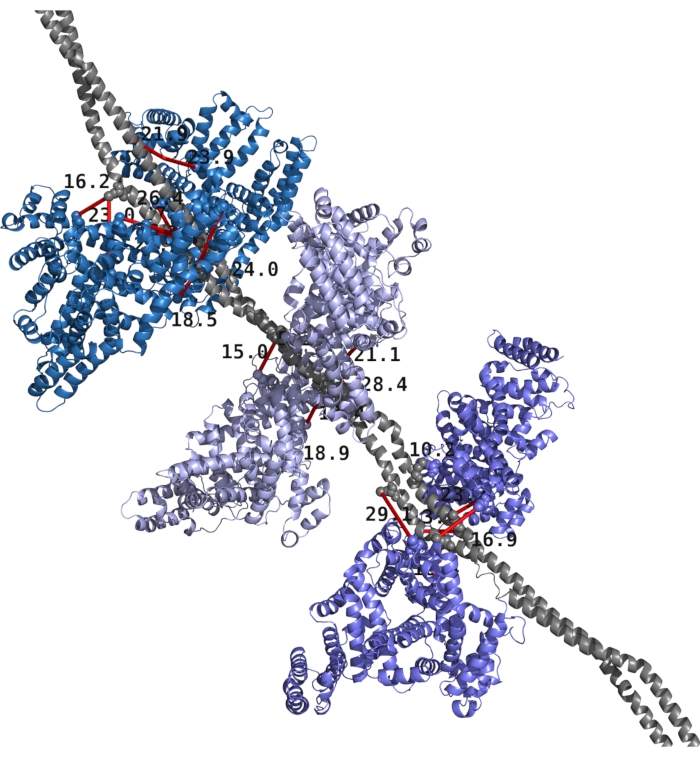
Figure 1: Structural model of Streptococcus pyogenes M1 protein and human albumin with XLs mapped on the structure. The M1 protein is shown in gray and constitutes a homodimer. The six albumin molecules are presented as pairs in various shades of blue. Cross-links and distances are given in red with black text. Please click here to view a larger version of this figure.
Supplementary File. Jupyter notebook data. Please click here to download this File.
Discussion
Modern computational workflows are often complex, with multiple tools from many different vendors, complex interdependencies, high data volumes, and multifaceted results. Consequently, it is increasingly difficult to accurately document all the steps required to obtain a result, making it difficult to reproduce the given result. Here, we demonstrate a general strategy that combines the automation and ease of an automated workflow that produces a generic report, with the flexibility to customize the report in a reproducible fashion.
Three requirements need to be fulfilled for the protocol to work: First, the proteins selected for analysis need to interact in such a manner that the chemical cross-linking experiment can produce cross-linked species at a sufficiently high concentration to be detected by the mass spectrometer; different mass spectrometers have different levels of detection and are also dependent on the acquisition protocol as well as the choice of cross-linking reagent. The current version of TX-MS protocol only allows for DSS, a lysine-lysine homobifunctional cross-linking reagent. Still, this limitation is primarily due to the possibility that the machine learning step would need to be adjusted for other reagents. This limitation has been improved in the Cheetah-MS web server as two more cross-linking reagents can be considered, but all three are non-cleavable reagents. Second, the two proteins need either to have an experimentally determined structure or be modeled using comparative modeling techniques or de novo techniques. Not all proteins can be modeled, but a combination of improved software and a constant deposition of experimental structures in the PDB expands the number of proteins that can be modeled. Third, the interacting proteins should remain sufficiently similar in their bound and unbound states so that the docking algorithms in use by TX-MS and Cheetah-MS can create quaternary structures of adequate quality to enable scoring. This requirement is relatively vague, as acceptable quality is highly system-dependent, where smaller proteins of known structure are generally easier to compare than larger proteins of unknown structure.
In case of a negative result, first check that TX-MS found intra-links, cross-links between residues that are part of the same polypeptide chain. If none are discovered, the most likely explanation is that something went wrong with the sample preparation or the data acquisition. If multiple distance constraints do not support the models, visually inspect the models to ensure that the conformation is supported by cross-linked residues. There is no obvious way to pivot one of the interactors without disrupting at least one cross-link. If there are cross-links longer than the permitted distance for the given cross-linking reagent, try to improve the modeling of the interactors by incorporating cross-linking data.
It is possible to use alternative software applications to accomplish equivalent results provided that the sensitivity of the chosen software is comparable to the sensitivity of TX-MS. For example, there are online versions of RosettaDock, HADDOCK, and others. It is also possible to analyze chemical cross-linking data through xQuest/xProphet5,6, plink7, and SIM-XL26.
We are continuously applying TX-MS and Cheetah-MS to new projects27,28,29, thereby improving the reports produced by these approaches to allow for a more detailed analysis of results without making the reports larger.
Divulgazioni
The authors have nothing to disclose.
Acknowledgements
This work was supported by the Foundation of Knut and Alice Wallenberg (grant no. 2016.0023) and the Swiss National Science Foundation (grant no. P2ZHP3_191289). In addition, we thank S3IT, University of Zurich, for its computational infrastructure and technical support.
Materials
| Two Protein DataBank files of the proteins of interest. | N/A | N/A | Example files available on txms.org and zenodo.org, DOI 10.5281/zenodo.3361621 |
| An mzML data file acquired on a sample where the proteins of interest were crosslinked. | N/A | N/A | Example files available on txms.org or zenodo.org, DOI 10.5281/zenodo.3361621 |
Riferimenti
- Berman, H. M., et al. The Protein Data Bank. Acta Crystallographica Section D: Biological Crystallography. 58 (6), 899-907 (2002).
- Herzog, F., et al. Structural Probing of a Protein Phosphatase 2A Network by Chemical Cross-Linking and Mass Spectrometry. Science. 337 (6100), 1348-1352 (2012).
- Hoopmann, M. R., et al. Kojak: efficient analysis of chemically cross-linked protein complexes. Journal of Proteome Research. 14 (5), 2190-2198 (2015).
- Seebacher, J., et al. Protein cross-linking analysis using mass spectrometry, isotope-coded cross-linkers, and integrated computational data processing. Journal of Proteome Research. 5 (9), 2270-2282 (2006).
- Rinner, O., et al. Identification of cross-linked peptides from large sequence databases. Nature Methods. 5 (4), 315-318 (2008).
- Walzthoeni, T., et al. False discovery rate estimation for cross-linked peptides identified by mass spectrometry. Nature Methods. 9 (9), 901-903 (2012).
- Yang, B., et al. Identification of cross-linked peptides from complex samples. Nature Methods. 9 (9), 904-906 (2012).
- Chu, F., Baker, P. R., Burlingame, A. L., Chalkley, R. J. Finding Chimeras: a Bioinformatics Strategy for Identification of Cross-linked Peptides. Molecular & Cellular Proteomics. 9 (1), 25-31 (2010).
- Holding, A. N., Lamers, M. H., Stephens, E., Skehel, J. M. Hekate: Software Suite for the Mass Spectrometric Analysis and Three-Dimensional Visualization of Cross-Linked Protein Samples. Journal of Proteome Research. 12 (12), 5923-5933 (2013).
- Hauri, S., et al. Rapid determination of quaternary protein structures in complex biological samples. Nature Communications. 10 (1), 192 (2019).
- Röst, H. L., et al. OpenSWATH enables automated, targeted analysis of data-independent acquisition MS data. Nature Biotechnology. 32 (3), 219-223 (2014).
- Röst, H. L., et al. OpenMS: a flexible open-source software platform for mass spectrometry data analysis. Nature Methods. 13 (9), 741-748 (2016).
- Quandt, A., et al. Using synthetic peptides to benchmark peptide identification software and search parameters for MS/MS data analysis. EuPA Open Proteomics. 5, 21-31 (2014).
- Bradley, P., et al. Free modeling with Rosetta in CASP6. Proteins: Structure, Function, and Bioinformatics. 61 (S7), 128-134 (2005).
- Gray, J. J. High-resolution protein-protein docking. Current Opinion in Structural Biology. 16 (2), 183-193 (2006).
- Khakzad, H., et al. Cheetah-MS: a web server to model protein complexes using tandem cross-linking mass spectrometry data. Bioinformatics. , (2021).
- Malmström, L. Chapter 15: Computational Proteomics with Jupyter and Python. Methods in Molecular Biology. 15, 237-248 (1977).
- Martens, L., et al. mzML–a community standard for mass spectrometry data. Molecular & Cellular Proteomics. 10 (1), (2011).
- Chambers, M. C., et al. A cross-platform toolkit for mass spectrometry and proteomics. Nature Biotechnology. 30 (10), 918-920 (2012).
- Waterhouse, A., et al. SWISS-MODEL: homology modelling of protein structures and complexes. Nucleic Acids Research. 46 (W1), W296-W303 (2018).
- Yang, J., et al. Improved protein structure prediction using predicted interresidue orientations. Proceedings of the National Academy of Sciences. 117 (3), 1496-1503 (2020).
- Koehler Leman, J., et al. Macromolecular modeling and design in Rosetta: recent methods and frameworks. Nature Methods. 17 (7), 665-680 (2020).
- Chivian, D., et al. Prediction of CASP6 structures using automated Robetta protocols. Proteins: Structure, Function, and Bioinformatics. 61 (S7), 157-166 (2005).
- Chivian, D., et al. Automated prediction of CASP-5 structures using the Robetta server. Proteins: Structure, Function, and Bioinformatics. 53 (S6), 524-533 (2003).
- Bauch, A., et al. openBIS: a flexible framework for managing and analyzing complex data in biology research. BMC Bioinformatics. 12, 468 (2011).
- Lima, D. B., et al. SIM-XL: A powerful and user-friendly tool for peptide cross-linking analysis. Journal of Proteomics. 129, 51-55 (2015).
- Happonen, L., et al. A quantitative Streptococcus pyogenes-human protein-protein interaction map reveals localization of opsonizing antibodies. Nature Communications. 10, 2727 (2019).
- Khakzad, H., et al. Structural determination of Streptococcus pyogenes M1 protein interactions with human immunoglobulin G using integrative structural biology. PLOS Computational Biology. 17 (1), E1008169 (2021).
- Khakzad, H., et al. In vivo cross-linking MS of the complement system MAC assembled on live Gram-positive bacteria. Frontiers in Genetics. 11, (2020).
