In this representative experiment, the BK-polyomavirus Non-Coding Control Region driven transcriptional activity was measured via flow cytometry. In addition, a mTOR inhibitor, which might be used to treat patients after BKPyV reactivation, was tested for its inhibition of the viral early gene expression. To this end, a dual fluorescence-reporter assay was used as published previously5. The overall workflow scheme of the experimental setup is illustrated in Figure 1. First, blood samples from immunocompromised renal transplanted patients were collected according to the guidelines of human research as approved by the institutional ethics committee. The blood was collected in EDTA-containing tubes and the phases were separated by centrifugation. Subsequently, 400 µL of plasma was used for isolation of the polyomavirus DNA. The non-coding control region (NCCR) was amplified using a nested-PCR protocol as illustrated in Figure 2A using the outer primer pair BKV-CR-1fw and BKV-CR-1rv, as well as, the inner primer pair BKV-CR-2fw-AgeI (Primer AgeI) and BKV-CR-2rv-SpeI (Primer SpeI)17, the latter of which harbors the restriction sites for AgeI and SpeI. The amplicon verification was performed via agarose gel electrophoresis as shown in Figure 2B. While the amplicons derived from archetypical NCCR sequences (wt) had a homogeneous size distribution in the gel, those derived from rearranged NCCRs with insertions (rrins) and deletions (rrdel) differed in their size (approx. 300-500 bp). Notably, due to rearrangements, the Dunlop NCCR reference sequence16 (DUN), which was included as a control, was larger than the NCCR obtained from archetypical viruses. Since the amplicons corresponded to the predicted sizes, the purified PCR products were sent for Sanger sequencing for later comparison with the sequences cloned into the reporter plasmid for further analysis.
For cloning of the amplicons, digestion was performed using the two restriction enzymes AgeI and SpeI. After cloning into the dual fluorescence reporter, which was performed using standard cloning procedure, selection of positive clones containing the NCCR was verified by AgeI and SpeI digestion (Figure 2C). Here, the small spacer region (NC, 128 bp) was replaced by the larger NCCR-fragments (approx. 300-500 bp) indicating positive clones. Plasmid DNA isolated from verified clones were sent for Sanger sequencing and compared with the sequences obtained from amplicon sequencing (not shown). Only the clones that match the amplicon sequence were chosen for further processing. As depicted in Figure 3, the sequencing results were compared with an archetypical NCCR sequence (JN192438). In this example, a deletion in the P-block and a substitution in O-block were detected.
To subsequently test the transcriptional activity of the cloned NCCR sequences in cell culture, HEK293T cells were transiently transfected with the respective reporter constructs (Figure 4A). As visualized in Figure 4B, the transfection efficiency and NCCR-activity were monitored via fluorescence microscopy. Red and green fluorescence corresponded to early and late BKPyV gene expression, respectively5. The cells expressed both fluorophores indicating efficient early and late promotor activity. As illustrated by the two examples, the first NCCR (NCCR1) displayed a strong late gene expression, while the second example (NCCR2) had a comparatively strong early but moderate late gene expression (Figure 4B). Thus, samples were prepared for the flow cytometry measurement. As described in protocol section 6, DAPI negative cells were gated (Figure 5A, lower panel) and a dot plot was created showing eGFP versus tdTomato. Samples that were negative (Figure 5B), as well as those positive for tdTomato (Figure 5C) or eGFP (Figure 5D) only, were included into the analysis. Note, initial compensation was performed, which is essential to distinguish red and green signals and to achieve accurate results18. Mean fluorescence intensities were derived from each testing clone. Here, tdTomato positive cells corresponded to early expression while eGFP positive cells represented late expression. In this example the treatment with the dual mTOR inhibitor INK128 significantly reduced tdTomato MFI (Figure 5E-F), which means that early expression was inhibited.
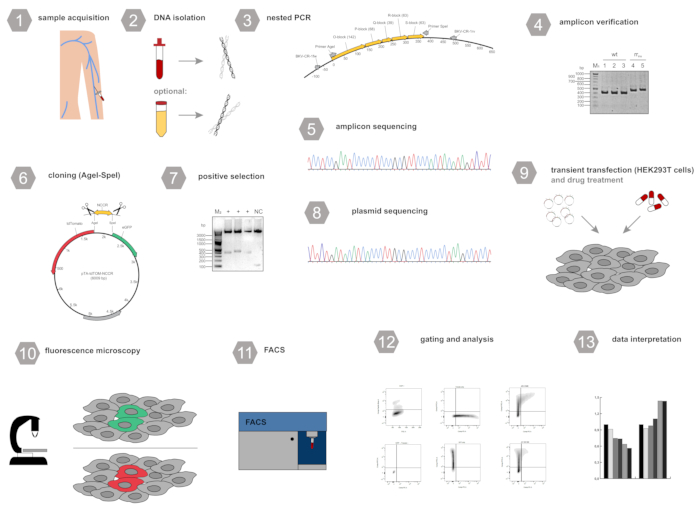
Figure 1: Workflow scheme for the experimental setup.1) Collection of blood (alternatively urine) samples, 2) Isolation of polyomavirus DNA 3) Amplification of the non-coding control region (NCCR) using a nested-PCR protocol, 4) Agarose gel electrophoresis and amplicon verification, 5) Sanger sequencing of the PCR amplicon, 6) Cloning of the NCCR into the dual fluorescence reporter using AgeI and SpeI, 7) Selection of positive clones, 8) Sanger sequencing of the plasmid DNA, 9) Transient transfection of HEK293T cells with the reporter plasmid and addition of compounds to be tested, 10) Fluorescence microscopy to verify efficient transfection, 11) Flow cytometry analysis, 12) Gating and analysis of eGFP tdTomato expression, 13) Data interpretation. Please click here to view a larger version of this figure.
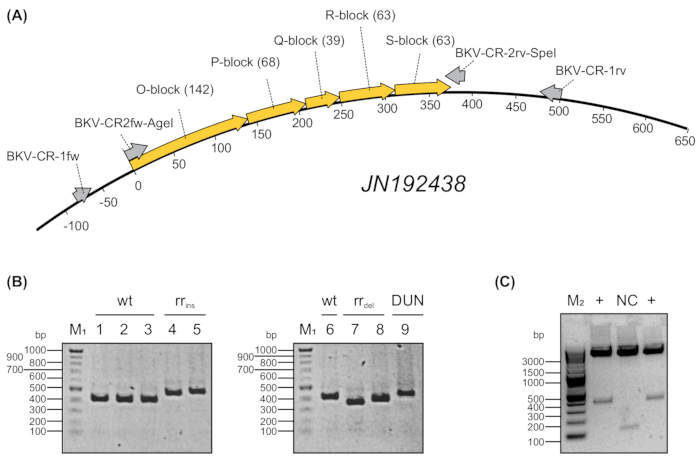
Figure 2: Nested PCR and representative analysis of patient derived NCCR-amplicons.A) DNA derived from immunocompromised renal transplantedpatients was subjected to semi-nested PCR amplification using oligonucleotide primers linked with restriction sites AgeI and SpeI. Primer names and length (base pairs) of archetypical O, P, Q, R, and S-blocks from BKPyV strain JN192438 are indicated in brackets. B) Amplicons were analyzed by electrophoresis in 1.5% agarose gel and visualized using DNA staining. M1 = 100 bp ladder, wt = wildtype (archetypical), rr = rearranged, ins = insertions, del= deletions, DUN = Dunlop Strain. C) Plasmid digestion with AgeI and SpeI. Digested fragments were analyzed by electrophoresis in 1.5% agarose gel and visualized using DNA staining. M1 = 100 bp ladder, M2 = 2-log ladder. NC = negative control (spacer). + = positive clone. Please click here to view a larger version of this figure.
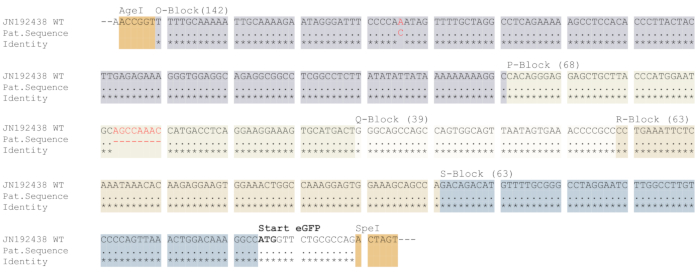
Figure 3: Cloning and validation of the NCCR into the dual fluorescence reporter. Representative results of plasmid sequencing. Amplicons were purified using a PCR purification kit and subjected to Sanger sequencing. The respective sequences were aligned to the NCCR sequence derived from archetypical BKPyV strain JN192438. Deletions and substitutions are marked in red. The AgeI and SpeI restriction sites, translational start of eGFP open reading frame (printed in bold) as well as the NCCR blocks O-S are indicated. The respective blocks and the restriction sites are highlighted in color. The length of each NCCR block in base pairs are printed in brackets. Please click here to view a larger version of this figure.

Figure 4: Representative fluorescence microscopy analysis of early and late NCCR promoter activity.A) Gene map of the dual fluorescence reporter under the control of the bidirectional promoter. As described previously5 the plasmid pTA-tdTOM-NCCR-eGFP (6,009 bp) harbors the SV40 late polyadenylation signal downstream of each expression cassette to ensure comparable and efficient processing of both transcripts for tdTomato (early expression) and eGFP (late expression), respectively. NCCR is flanked by AgeI and SpeI. The vector contains an ampicillin resistance cassette for selection during cloning procedure. B) HEK293T cells were transfected with dual-fluorescence reporter constructs each under the control of patient derived NCCR sequences (NCCR1-2). Cells were subjected to brightfield and fluorescence microscopy analysis 72 h post transfection. Filter settings of the microscope were used in the following excitation ranges: green for tdTomato (515-560 nm) and blue for eGFP (450-490 nm). The scale bar (200 µm) is indicated in the bottom right of each photography. Please click here to view a larger version of this figure.
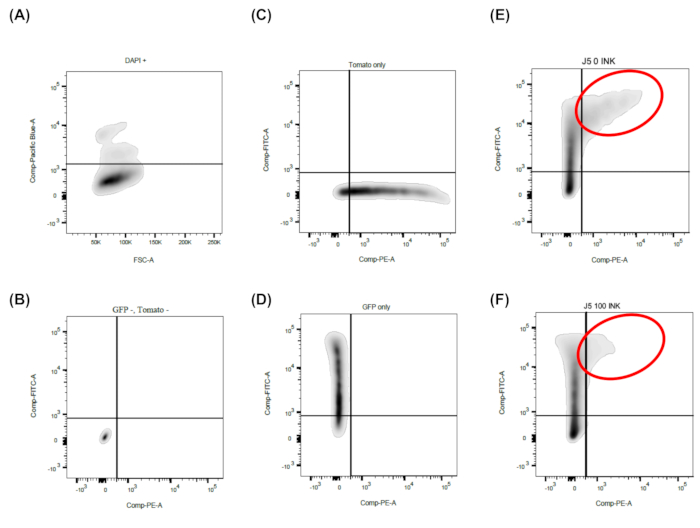
Figure 5: Representative FACS analysis. Shown is the simple gating strategy required for this method. Two-parameter density or dot plots are used. A) First, FSC is plotted against Pacific Blue, while only DAPI negative (i.e., living cells) are further processed. B-F) FITC (eGFP) versus PE (tdTomato) are plotted. C-D) Add exclusively green and red fluorescent cells to adjust the compensation on the flow cytometer. E-F) In this example, the mTOR inhibitors INK128 and significantly reduces the tdTomato MFI, which corresponds to a reduction of the BKPyV early gene expression. Please click here to view a larger version of this figure.
| Primer name | Sequence (5' → 3') |
| BK-CR-fwd1 | CCCAGGCAGCTCTTTCAAGGC |
| BK-CR-rev1 | CCTCTAACAAAATTCCAGCAAAAGC |
| BKV-CR-2-fw-AgeI | AAAAAAACCGGTTTTTGCAAAAATTGCAAAAGAATAGG |
| BKV-CR-2-rv-SpeI | TTTTTTACTAGTCTGGCGCAGAACCATGGCCTT |
| EGFP-N | CGTCGCCGTCCAGCTCGACCAG |
Table 1: Oligonucleotides used in this protocol. The underlined bases indicate the restriction enzyme sites for AgeI and SpeI, respectively.
| Component | Volume/reaction (µl) | final concentration |
| RNase-free water | 32.8 µL | – |
| 10x PCR Buffer | 5 µL | 1x |
| dNTPs (10 mM of each) | 1 µL | 0.2 mM of each dNTP |
| Fwd-primer (10 µM) | 3 µL | 0.3 µM |
| Rev-primer (10 µM) | 3 µL | 0.3 µM |
| Taq Polymerase | 0.2 µL | 2 unit/reaction |
| Template DNA | 5 µL | <0.5 µg/50 µL reaction |
Table 2: Master mix preparation protocol. The instruction applies to both pre- and nested PCR reactions as described in steps 2.2 and 2.7, respectively.
| Temperature | Duration | PCR-step | cycles |
| 95°C | 5 min | Initial Denaturation | |
| 95°C | 30 sec | Denaturation | |
| 55°C | 34 sec | Annealing | x35 |
| 72°C | 1 min | Extension | |
| 72°C | 10 min | Final extension | |
| 4°C | ∞ | Cooling |
Table 3: PCR program used for NCCR amplification. The instruction applies to both pre- and nested PCR reactions.
| Reagent | Volume/reaction (µl) | final concentration |
| DNase-free H20 | 6 | to 20 µl |
| 10x Buffer | 2 | 1x |
| AgeI | 1 | 10 units |
| SpeI | 1 | 10 units |
| purified PCR-amplicon | 10 | variable |
Table 4: Amplicon digestion protocol. The instruction applies for the simultaneous digestion of the amplicon DNA with two restriction enzymes described in step 3.1.
| Reagent | Volume/reaction (µl) | final concentration |
| DNase-free H20 | 14.5 | to 20 µl |
| 10x Buffer | 2 | 1x |
| AgeI | 1 | 10 units |
| SpeI | 1 | 10 units |
| Plasmid backbone (1 µg/µL) | 1.5 | 1.5 µg |
Table 5: Plasmid digestion protocol. The instruction applies for the simultaneous digestion of the plasmid DNA backbone with two restriction enzymes described in step 3.3.
| Reagent | Volume/reaction (µl) | final concentration |
| DNase-free H20 | 7 | to 20 µl |
| T4 DNA ligase | 1 | 20 |
| 10x T4 DNA ligase buffer | 2 | 1x |
| Purified Plasmid backbone Low melt diluted ½ from step 3.5 |
2 | variable |
| Digested and purified PCR-amplicon from step 2.7 | 8 | variable |
Table 6: Plasmid ligation protocol. The instruction applies for the ligation of the plasmid DNA backbone with the digested PCR amplicon DNA described in step 3.7.
