The study was carried out in accordance with European Community guidelines on the care and use of laboratory animals: 86/609/EEC. The research purpose and the protocol are described in the Ethical Annex of ERCadg project CellO, which was approved and is regularly reviewed by the ERCEA. Institut Curie animal facility has received licence #C75-05-18, 24/04/2012, reporting to Comité d'Ethique en matière d'expérimentation animale Paris Centre et Sud (National registration number: #59).
1. Fabrication of the mold
NOTE: The mold includes central and intermediate chambers connected to an inlet and an outlet, plus two reservoirs (and inlets) located on both sides of the central chamber.
- Use flat tweezers to manipulate 51 mm diameter silicon wafers and transfer them from one place to another.
- Central chamber silicon mold
- Fill a 2-mL plastic pipette with positive photoresist. Position the pipette at the center of a 51-mm diameter silicon wafer and press on its reservoir until covering about 75% of the wafer surface with the photoresist. Spin-coat at 3000 rpm for 30 s.
- Transfer the wafer from the spin-coater to a hotplate, with a surface temperature of 100 °C, for 50 s.
- Transfer the wafer from the hotplate to the substrate holder (chuck) of the mask aligner. Expose through the "DRIE mask" (see Supplementary files "masks_neuron_volume_chips.tiff" and "masks_neuron_volume_chips.dxf").
- Remove the wafer from the chuck then dive it in a 100-mL glass crystallizing dish containing the developer (dilution 1:4 in deionized water). Release the wafer and maintain it for 1 min while continuously and gently stirring the crystallizing dish.
NOTE: To save reagents, fill the crystallizing dish to 1 cm in height at maximum. - Take back the wafer from the developer and submerge it into a 100-mL crystallizing dish filled with deionized water for about 10 s.Then place the wafer on an absorbent paper and dry it with pressurized nitrogen using an air blow gun.
- Place the wafer for 50 s on a hotplate with a surface temperature of 115 °C.
- Perform deep-Reactive Ion Etching (DRIE) with the following parameters (more information on the DRIE technique can be found in references 14 and 15); Passivation step: 50 sccm of C4F8, He backing flow 10 sccm, CP 10 W, Inductively Coupled Plasma (ICP) 1500 W, total pressure 24 mbar, temperature 18 °C; Etching step: 100 sccm of SF6, He backing flow 10 sccm, CP 11 W, ICP 1500 W, total pressure 24 mbar, temperature 18 °C; Passivation time: 4 s; Etching time: 7 s; Total process duration: typically 5 min for about 10 µm etched depth.
- Dissolve the photoresist by diving the substrate into a crystallizing dish filled with acetone.
- Submerge the wafer in a piranha solution (2/3 of H2O2 (30%) and 1/3 of H2SO4 (95%)) for 5 min.
Caution: Always add H2O2 first and then H2SO4 and rinse at least 3 times in deionized water after cleaning.
- Fabricate the molds corresponding to intermediate chambers by performing steps 1.2.1 to 1.2.11 following the parameters listed in Tables 1-3.
NOTE: Each table corresponds to a given device characterized by a specific height of the central observation chamber.
NOTE: Perform the whole process using increasing photoresists heights (Masks 1 to 3 for each device, see Supplementary files "masks_neuron_volume_chips.tiff" and "masks_neuron_volume_chips.dxf").- Place the etched silicon wafer on the spin-coater substrate holder.
- Check that the spin-coater is working properly by verifying that the aspiration of the substrate is effective (the substrate should spin and stay in place during the nominal rotation speed).
- The viscosity of SU-8 increases with the height of the targeted thickness range. Always use 20-30 mL bottles to store SU-8 photoresists and pour it on the wafer before spin-coating (SU-8 might be too viscous to be manipulated with a plastic pipette).
- Pour negative epoxy-type photoresist SU-8 at the center of the substrate until covering about 75% of the silicon wafer, then spin-coat using the parameters indicated in row "Spincoating" of the Tables.
- Place the coated wafer on a hotplate for the duration and temperature indicated in row "Soft bake".
- Mount the etched wafer and the adequate "SU-8 Mask" on the mask aligner.
- Align the etched wafer with the mask using the dedicated alignment crosses (typical size of these crosses: 50 µm × 150 µm) designed on each mask.
NOTE: Two crosses on each side of the device are sufficient (one at the bottom left and one at the top right). - Expose using the I-line of the mask aligner (wavelength 365 nm) with the appropriate UV dose, as indicated in row "Exposure energy".
NOTE: The exposure time is calculated by dividing the exposure energy E specific for each photoresist by the effective power of the UV lamp, modulated by the absorption of the mask: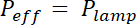 x (1- Absorptionmask). Absorption is about 20% for flexible masks and negligible for chromium hard mask.
x (1- Absorptionmask). Absorption is about 20% for flexible masks and negligible for chromium hard mask. - Place the coated wafer on a hotplate for the duration and temperature indicated in row "Post bake".
- Prepare two 100-mL glass-crystallizing dishes, one containing the developer, the other empty. Dive the wafer in the developer for the duration indicated in row "Development". Agitate gently the crystallizing dish all along development
- Sprinkle the wafer with isopropanol above the empty crystallizing dish for about 5 s. Finally, place the wafer on an absorbent paper and dry it with pressurized nitrogen using an air blow gun.
- Place the coated wafer on a hotplate for the duration and temperature indicated in row "Hard bake" (optional).
NOTE: This step is useful to avoid cracks in the photoresist and provide a homogeneous flat surface for the next steps. - Repeat steps 1.3.1 – 1.3.10 to complete the processes listed in Table 1-3.
- After the last step of photolithography, silanize the final master mold consisting of 3 layers of negative photoresists by dispatching two 100 µL droplets of Trichloro(1H,1H,2H,2H-perfluoro-octyl) silane on each side of the master in a 100-mm Petri dish. Seal the Petri disk with a plastic paraffin film and incubate 20 min at room temperature (RT).
NOTE: The master mold is ready and can be used several times.
- Place the etched silicon wafer on the spin-coater substrate holder.
2. Fabrication of PDMS chip
- Pour 90 g of silicon-based organic polymer (Polydimethylsiloxane: PDMS) into a 100-mL plastic cup. Add 10 g of its curing agent (1:10 in weight). Stir the mixture using a plastic pipette for 2-3 min.
NOTE: Mix 90 g of PDMS with 10 g of curing agent for the fabrication of 6 chips. - Place the mixture within a vacuum desiccator and pump for about 30 min to remove air bubbles trapped into the PDMS.
- Place the master mold in a P100 Petri dish and pour 15 mL of the mixture on the mold using a syringe.
NOTE: 15 mL leads to a total chip height of 1.5 mm. - Place the PDMS-mold structure within a vacuum desiccator and pump until there are no more air bubbles bursting at the surface of the PDMS.
NOTE: This step takes about 30 min. - Push the wafer at the bottom of the Petri dish using a cone tip to avoid dead PDMS volume below the wafer. Place the PDMS-mold device in the oven set at 70 °C for at least 2 h.
- Demold the PDMS block under a chemical hood using a stainless steel flat spatula and by pouring isopropanol on the chip. Place it on the stainless steel bench of the hood.
- Cut around the silicon wafer and demold the PDMS replica using a scalpel.
- Cut around (leave a 2-mm margin) the chip using a scalpel or a razor blade.
- Punch inlets by pressing the 1.5 mm diameter puncher firmly and actuate it to cut and carve the hole of the inlet. Do the same at the four dedicated zones of the chip where liquid will be injected.
- Clean the chip by sticking and peeling adhesive tape on the microstructured side. Sprinkle isopropanol on both sides. Then dry the chip with pressurized nitrogen using an air blow gun.
3. Fabrication of patterned coverslips (24 × 24 mm2)
NOTE: Manipulate coverslips with curved tweezers.
- Poly-ornithine (PLO) patterns
- Apply an O2 cleaning plasma on glass coverslips. Plasma parameters: pumping down pressure: 0.25 mbar; O2 supply duration: 3 min; gas flow: 10 sccm; maximum deviation: ±5 sccm; plasma duration: 3 min; set pressure: 0.36 mbar; maximum deviation: ±0.10 mbar; set power: 50 W; maximum deviation: 5%; venting duration: 45 s.
- Mix 484 µL of acetic acid and 56 µL of 3-methacryloxypropyl-trimethoxysilane, complete with absolute ethanol to get a total volume of 15 mL.
- Using a 1-mL tip cone, put a drop of 500 µL of this solution on each coverslip, wait for 2-3 min. Dry using a cleanroom microfiber wiper.
NOTE: Silanized glass coverslips can be stored up to 1 month at room temperature within plastic boxes sealed with a plastic paraffin film. - Place each coverslip on a spin coater located in a clean room environment. Put a drop of a positive photoresist covering about 75% of the coverslip (about 500 µL) and spin-coat at 4000 rpm for 30 s to reach a final thickness of 0.45 µm.
- Place the coverslips for 1 min on a hotplate with a surface temperature of 115 °C.
- Using a mask aligner, expose each coverslip at a wavelength of 435 nm (G-line) through the dedicated mask according to the fabricant parameters (UV dose about 50-60 mJ.cm–1)
- Prepare 2 glass crystallizing dishes, one containing the developer (no dilution), the other containing deionized water.
- Dive one by one each coverslip in the developer for 1 min while continuously and gently stirring the crystallizing dish. Then submerge the wafer in deionized water for about 5 s. Place the wafer on an absorbent paper and dry it with pressurized nitrogen using an air blow gun.
- Apply an activation O2 plasma with the same parameters as in 3.1.1.
- Under the hood, depose four 170 µL drops of a 100 µg/mL PLO solution per P100 Petri dish. Put the patterned face of the coverslips on each of these drops. Seal the Petri dish with a plastic paraffin film to avoid drying. Incubate overnight at RT.
NOTE: The PLO solution should remain attached to the coverslips by capillarity. - Prepare four recipients (typically P60 Petri dishes), fill three of them with PBS and the fourth one with deionized water. Prepare two glass crystallizing dishes of pure ethanol.
- Take out each coverslip from the Petri dishes, submerge it in the first PBS bath for 10-15 s, evacuate the liquid on an absorbing tissue by putting the coverslip vertically on the side, insert it with e.g. the patterned face up inside the ethanol bath.
NOTE: Once the dissolution of the photoresist is completed, it becomes difficult to locate the patterned side of the coverslip. Therefore, it is important at this stage to trace its location. - Place the ethanol crystallizing dish within an ultrasonic bath sonicator (120 W / 35 kHz) and let the photoresist be dissolved for 3 min.
NOTE: Change the ethanol bath every 4 coverslips to limit dilution by the PBS that might impair the dissolution of the photoresist. - Take out the coverslip from the ethanol bath, then dive it several times into the second PBS bath. Check the surface aspect repeatedly until the greasy-like surface that results from the remaining liquid film of ethanol disappears.
- Submerge for 5-10 s the coverslip into the third PBS bath. Then immediately transfer it to the deionized water bath. Place the coverslip on an absorbent paper and dry it with pressurized nitrogen using an air blow gun.
NOTE: The last rinse in deionized water is used to avoid the formation of PBS crystals during the drying step.
- Photoresist structures for height calibration
- Perform only steps 3.1.4. to 3.1.8. using the dedicated mask (mask "Photoresist stripes", see Supplementary file "Mask_Photoresist-stripes.dxf").
4. Chip assembling and final implementation
- Chip assembling on glass bottom dishes
- Put both the PDMS chip and the glass dish on which it will be bonded into the plasma chamber for surface activation. Parameters: pumping down pressure: 0.25 mbar; O2 supply duration: 3 min; gas flow: 10 sccm; maximum deviation: ±5 sccm; plasma duration: 30 s; set pressure: 0.40 mbar; maximum deviation: ±0.10 mbar; set power: 50 W; maximum deviation: 5%; venting duration: 45 s.
- Gently put the activated PDMS chip in contact with the glass coverslip, and delicately apply pressure on the edges of the chip to bond the chip to the coverslip. To increase the bonding strength, place the device in the oven at 70 °C for 5 to 10 min.
NOTE: Do not press on parts containing pillars, they might collapse under too much pressure. - Under the hood (i.e. at RT) and within 30 min after bonding, place a 10-µL tip cone filled with a 100 µg/mL PLO solution at the IHs, then inject the liquid. Adjust the volume in order to form a drop at the top of each IHs. Then, using a 1-mL tip cone, add PBS in the Petri dish all around the chip.
- Let the chip at RT with a minimum of 2 h incubation time. For overnight incubation, seal the Petri dish using a plastic paraffin film to avoid drying.
NOTE: The liquid should not leak outside the chip otherwise the chip must be discarded. - Press a 10-µL tip cone lightly into each IH and suck up the excess of liquid. Then stick completely the tip cone inside the outlet and draw up the remaining liquid.
- Replace PLO by laminin following the instructions given in steps 4.1.5 (emptying) and 4.1.3 (filling). Incubate at RT for 1 h.
- Replace laminin by the culture medium following the instructions given in steps 4.1.5 (emptying) and 4.1.3 (filling). Composition of the culture medium: MEM 81.8%; Sodium Pyruvate 100 mM 1%; Glutamax 200 mM 1%; Horse Serum 5%; B27 supplement 2%, N2 supplement 1%, Gentamicin 0.2%; filter the solution using a 220-nm filter. Using a 1-mL tip cone, replace also the PBS surrounding the chip by this medium.
- Put the chip into the incubator regulated at 37 °C and 5% CO2 for at least 5 h (or overnight) before neuron seeding.
- Chip assembling using patterned coverslips
NOTE: If the patterned coverslips include positive photoresist reference objects, perform steps 4.2.1 to 4.2.9. Otherwise, perform only step 4.2.3, stick the PDMS device on the PLO patterned coverslip as indicated in 4.1.2, put culture medium inside and around the chip then go to step 4.2.10.- Deposit a drop of water on a rectangular thick microscope glass slide and stick the coverslip on the glass slide by capillarity (non-patterned side facing the glass slide). Under a microscope, mark the location of the photoresist stripes at the rear of the glass slide using a felt pen.
- Place the patterned glass coverslip on the mask holder of the mask aligner. Rely on the mark made with the felt pen to center the photoresist reference objects.
- Perform the plasma step as described in 4.1.1 on the PDMS chip.
- Place the PDMS chip on the mobile substrate holder (chuck) of the mask aligner.
NOTE: To increase optic contrast, place a silicon wafer beneath the PDMS chip. The silicon wafer should remain firmly attached on the chuck during the alignment process (use a transparent tape to stick it to the chuck). - Lift the chuck at the limit of mechanical contact to align the chip with the array of photoresist stripes located on the coverslip.
- Achieve mechanical contact between the chip and the coverslip by finishing up lifting the chuck until the surface of the PDMS pillars touch the glass coverslip.
- Lower the chuck. Remove the coverslip now bonded to the chip from the mask holder. Then place the device into a 35-mm Petri dish, and transfer everything into the oven (temperature: 70 °C) for 5 to 10 min to increase the bonding strength.
- Perform as in step 4.1.3.
NOTE: if using PLO patterned coverslips, go directly from step 4.2.7 to step 4.2.9. - Replace PLO by the plating medium following the procedures described in steps 4.1.5 (emptying) and 4.1.3 (filling). Using a 1-mL tip cone, replace also the PBS surrounding the chip by this medium.
- Put the chip into the incubator regulated at 37 °C and 5% CO2 until neuron seeding, with a minimum of 5 h incubation time.
5. Neuron culture
- Prepare 100 mL of dissection medium (HH medium) by mixing 10 mL of HBSS 10x, 2 mL of Hepes 1 M and 88 mL of sterile water in a 200-mL plastic flask.
NOTE: The HH medium can be prepared the day before the culture. - Dissect hippocampus from E18 mice embryo extracted from a mother euthanized by cervical dislocation (C57BL/6J mice from Charles Rivers). Steps of dissection are e.g. detailed in 16.
- Place hippocampus in a plastic tube containing trypsin (0.3 mL of trypsin 2.5%, w/o EDTA into 2.7 mL of HH medium) for 10 min at 37 °C in order to initiate chemical dissociation.
- Discard almost all the liquid and replace it with 5 to 10 mL of HH using disposable plastic pipettes. Do it 3 times. For the last fill, use 1 mL of plating medium instead of HH.
- Mechanically dissociate tissues using a 1 mL tip-cone by aspiring and ejecting the full volume several times, avoiding making bubbles and using no more than 15-20 passages.
- In a separate 500 µL recipient, prepare a solution using 5 µL of cell suspension diluted in 45 µL of PBS. Take 1 µL of this solution using a 10 µL pipette and insert the diluted suspension into a Malassez cell counter. Use the indications provided in 17 to estimate the number of cells.
NOTE: A single hippocampus usually provides about 0.5 million of neurons. - Centrifuge at 100 x g for 6 min at RT.
- Discard the supernatant and replace it by the volume of plating medium required to achieve a concentration of 10 million cells/mL. Resuspend cells by successively aspire and eject the cell suspension with a 1-mL tip cone.
- Draw up the plating medium present in the chip (refer to step 4.1.5). Collect 2-3 µL of the freshly resuspended solution using a 10-µL pipette and inject it at the inlet (refer to the injection procedure described in step 4.1.3). Repeat immediately the same operation at the outlet.
- Inject about the same volume of plating medium in each reservoir (refer to step 4.1.3).
NOTE: Quickly observe the chip under the microscope to check the density of cells. The order of magnitude of the optimal cell density corresponds to about 5-10 cells within the square surface delimited by 4 pillars (about 0.3 mm2, i.e. about 15-20 cells per mm2). - Eventually repeat step 5.10 using instead 0.5-1 µL of cell suspension to reach the targeted cell density.
- Place the seeded chip into an incubator regulated at 37 °C 5% CO2.
6. Fluorescence exclusion observation
- Replacement of culture medium by the imaging medium.
- Prepare the imaging medium as in 4.1.7 but use instead MEM devoid of phenol red and add fluorescent dextran. In that aim, dilute dextran (molecular weight 10,000 g/mol, stock solution concentrated at 10 mg/mL in PBS) in order to achieve a final concentration of 0.5-1 mg/mL in the imaging medium.
NOTE: Use Dextran conjugates with Absorption/Emission maxima 496/524 or 650/668. Prefer the first to image 0.45 µm high positive photoresist structures (to get rid of their auto fluorescence in the red bandwidth) and the second to image neurons (less toxic). - Empty all inlets using a 10-µL pipette and re-fill them completely with the imaging medium (refer to steps 4.1.3 and 4.1.5 for the precise methodology of medium replacement).
- Prepare the imaging medium as in 4.1.7 but use instead MEM devoid of phenol red and add fluorescent dextran. In that aim, dilute dextran (molecular weight 10,000 g/mol, stock solution concentrated at 10 mg/mL in PBS) in order to achieve a final concentration of 0.5-1 mg/mL in the imaging medium.
- Imaging
- Place the chip under an epifluorescence microscope equipped with an environmental chamber regulated at 37 °C and 5% of CO2. Use a 40X, numerical aperture (NA) 0.8 dry objective, 30% of full power (full power: 3 W) and 30 ms of exposure time. Acquire images of cells at focus (from single to multiple successive images in case of time-lapse experiments).
- Image analysis
- Normalize images using the background reduction routine implemented in dedicated software to get a homogeneous background. See 13 for the details of the image processing steps included in this software. The output image has a .MAT format.
- Convert .MAT file into .TIFF 8 bits images using the routine put in supplementary material (conversion_mattotiff.m, which calls for importfilevol.m).
- Perform Import > Image Sequence on ImageJ to build a video from the .TIFF images.
- Compute the mean intensity P of a square area localized in the center of a pillar (reference object) and the mean intensity B of a square area of the chamber devoid of cells (background, i.e. height zero). See 18 for an example of detailed methodology of image processing.
NOTE: The lateral dimension of the square areas used as intensity references must be about half of the pillar diameter to get a sufficient number of pixels while avoiding light pollution from the pillar edges. - Use the P and B values to establish the linear conversion law from intensity i to height h:
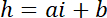
with hc the known height of the chamber,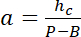 and
and 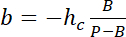
NOTE: PDMS displays no detectable autofluorescence. - Select an area around the zone of interest, integrate intensity using ImageJ (see 19 for more details) and apply the conversion law obtained in 6.3.5 to measure the volume of a cell compartment.
NOTE: The zone of interest might be selected based on the fluorescence of sub-cellular elements like actin, e.g., for the selection of growth cones in GFP-LifeAct neurons, select the zone of interest in the GFP emission channel, save the contour of this zone using a ROI manager tool, then measure the cell volume enclosed within the same zone in the emission channel of dextran (in red).
The result of the process of fabrication described in sections 1 and 2 is illustrated by the images of Figure 1A-1B and the curve of Figure 1C. The table of Figure 1D displays the roughness values of two different representative areas of the PDMS chip, i.e. in the central and the 20 µm high next intermediate chamber. A decrease in roughness by a factor of about 7 has been obtained by using etched Si wafer instead of SU-8 photoresist. Then, FXm was first applied on a photoresist stripe of known geometry (Figure 2A) within a 10 µm high chamber. After image processing and intensity to height conversion (see the graph of Figure 2B), FXm profiles performed on cross-sections along this stripe (Figure 2C) provide the desired height profiles (Figure 2D). Figure 2D shows the comparison between profiles obtained using mechanical profilometry and FXm methods. These profiles, including edge and plateau value, are very similar, validating the method. Note that the scattering of FXm data is not representative of the ultimate resolution of the method, as further assessed in Figure 3 and Figure 4, but results from the low intensity employed to avoid a possible effect of the very weak auto-fluorescence of the photoresist in the GFP channel.
Then, we observed neurites in 3 µm and 10 µm high chambers (Figure 3). The standard deviation of the background noise is about 18 nm after intensity to height conversion and background correction. This value is slightly higher than the physical roughness of PDMS surfaces casted on silicon surfaces (12 nm, see the table of Figure 1D) but much lower than the roughness measured on PDMS obtained from SU-8 molds. These results highlight the added value of drilling wells into silicon wafers rather than opening holes into SU-8 photoresist to cast pillars. Such a low value allows a high signal to noise ratio and very clear images in volume such as the one displayed in Figure 3A. As an example of the data that can be retrieved from such images, we computed the volume of 1.6 µm (i.e. 10 pixels) wide neurite slice (see the graph of Figure 3B). Using in a first approximation a linear fit of these data gives a mean neurite height value of about 400 nm, to be compared with e.g. the 500-nm axonal diameter found in 10 days old pups within the corpus callosum 5. We also combined FXm with micropatterns of adhesion consisting of serially abutted 2 µm and 6 µm wide stripes of 30 µm in length. Our aim was to study the influence of the neurite width on its 3D shape. Figure 3C shows a 3D representation in false color of a whole neuron image obtained in a 10 µm high chamber. Neurites are spreading on 2 µm and 6 µm wide stripes, whereas the soma is located on the extremity of the largest stripe. Height profiles were drawn in three different cross sections. In coherence with the graph displayed in Figure 3A, the surface integrated over the cross-sections increase with the neurite width (Figure 3D).
We also focused on growth cone (GC) 3D structures. Figure 4A-B displays two different GC profiles obtained in a 3 µm high chamber, which highlight their branched sub-structure. In addition, we performed time-lapse experiments to follow the dynamics of the volume of GCs in a 12 µm high chamber. Figure 4C displays a cycle of shrinking and reactivation of a given GC within a time scale of a few tens of minutes. Thanks to the use of GFP-lifeact mice, growth cones were localized in the GFP emission wavelength (510 nm) from their high actin concentration. The surface identified at the wavelength was used to integrate over the dextran emission wavelength at 647 nm to compute GC volume. Figure 4D shows finally the distribution of GC volume at different time points and location on three different neurons, centered on a value of about 6 µm3.
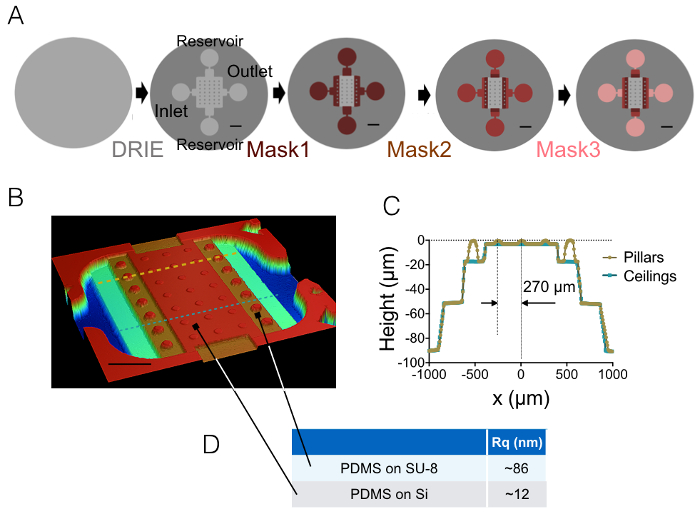
Figure 1: FXm PDMS chambers. (A) Schemes of the four different main steps of microfabrication leading to the final mold. The location of the inlet, outlet and reservoirs are indicated. Scale bars: 1 mm. (B) Image of the PDMS FXm chamber obtained using an optical profilometer. This image shows the central chamber containing 3 rows of 10 µm high pillars and the intermediate chambers of 20, 50 and 90 µm in height. Scale bar: 500 µm. (C) Cross-sectional view of the chip along the two dashed lines drawn in (B). Yellow/gold: cross-section along pillars, blue: cross-section between pillars. (D) Mean values of the PDMS roughness measured on 50 × 50 µm2 areas molded on silicon and on the 20 µm high SU-8 intermediate chamber (see arrows for the location of these areas). Mean values were obtained from the measurements of three different areas. Please click here to view a larger version of this figure.
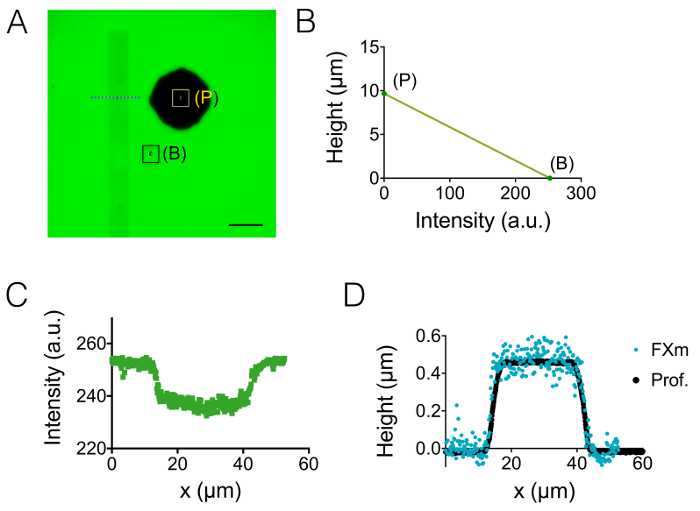
Figure 2: Calibration of FXm method using a photoresist stripe as the object of interest. (A) GFP-fluorescence image taken in a 10 µm high chamber filled with 10,000 MW dextran absorbing at 488 nm at 1 mg/mL. (B: background, P: pillar). Observation with a dry 40X NA 0.8 objective. Scale bar: 50 µm. (B) Linear calibration law obtained from the mean intensity of the two colored rectangles shown in A. (C) Fluorescence intensity profile obtained at the level of the blue dashed line displayed in (A), crossing the photoresist stripe (0.45 µm high positive photoresist). (D) Comparison of the profiles obtained from mechanical profilometer (black dots) and FXm after intensity to height conversion of the data of (B) (blue dots). Please click here to view a larger version of this figure.
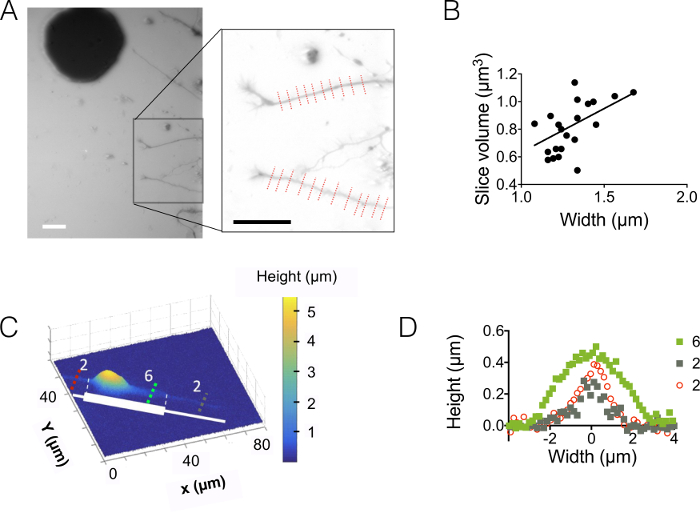
Figure 3: Neurite volume imaging. (A) Neurite extending into the central 3 µm high chamber from soma located in the next 15 µm intermediate chamber. Imaging performed using 10,000 MW dextran absorbing at 488 nm and a 40X, NA 0.8 dry objective. The inset obtained after the use of the background reduction routine highlights the two neurites and chosen to plot the graph on the right. Scale bars: 30 µm. (B) Neurite slice volume as a function of neurite width obtained from the 22 profiles (average on 10 pixels, i.e. on a 1.6 µm "neurite slice") shown in (A). The solid line represents a linear fit of slope 0.4 µm passing through the origin. (C) False color image of a patterned neuron on an adhesive stripe made of successive 2 µm and 6 µm wide stumps (represented in white). Measurements were made in a 10 µm high chamber filled with 10,000 MW dextran absorbing at 647 nm and using a 40x NA 0.8 dry objective. (D) Height profiles corresponding to the colored dashed lines shown in (C), keeping the same color code. Please click here to view a larger version of this figure.
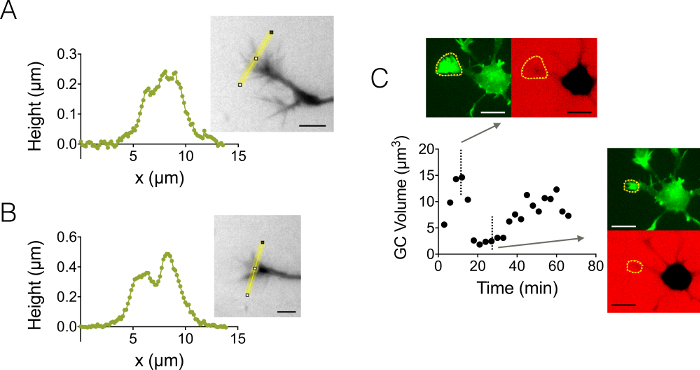
Figure 4: Static and dynamic growth cone imaging. (A-B) Growth cone height profiles obtained in a 3 µm high chamber after intensity to height conversion along the yellow lines displayed in associated images. Observation performed using a fill with 10,000 MW dextran absorbing at 488 nm and a 40X, NA 0.8 dry objective. (C) Whole neuron imaging in a 12 µm high chamber filled with 10,000 MW dextran absorbing at 647 nm. Observations have been made in two fluorescent channels: GFP for growth cone localization (dashed yellow lines), and CY5 to compute GC volume from fluorescence exclusion. The surface included by dashed yellow lines was used to compute GC volume. The graph shows the variation of GC volume over time, and associated morphologies in both GFP and CY5 channels at two representative different time points. All data were acquired using a 40x NA 0.8 dry objective every 3 min. Scale bars: 10 µm. Please click here to view a larger version of this figure.
| Step | Mask 1:8 µm layer | Mask 2:30 µm layer | Mask 3:40 µm layer |
| SU-8 type | 2007 | 2025 | 2050 |
| Spincoating | 30 s @ 2000 rpm | 30 s @ 3050 rpm | 30 s @ 3250 rpm |
| Soft bake | 3 min @ 95 °C | 2 min @ 65 °C + 6 min @ 95 °C | 3 min @ 65 °C + 7 min @ 95 °C |
| Exposure energy | 110 mJ/cm2 | 155 mJ/cm2 | 170 mJ/cm2 |
| Post-exposure bake | 4 min @ 95 °C | 1 min @ 65 °C + 6 min @ 95 °C | 2 min @ 65 °C + 7 min @ 95 °C |
| Development | 2 min 30 s | 5 min | 6 min |
| Hard bake (optional) | 3-5 min @ 200 °C | 3-5 min @ 200 °C | 3-5 min @ 200 °C |
Table 1: Photolithography steps performed to build a device containing a central chamber of 12 μm in height. Heights of the intermediate chambers: 20, 50 and 90 µm.
| Step | Mask 1:10 µm layer | Mask 2:30 µm layer | Mask 3:40 µm layer |
| SU-8 type | 2007 | 2025 | 2050 |
| Spincoating | 30 s @ 1500 rpm | 30 s @ 3050 rpm | 30 s @ 3250 rpm |
| Soft bake | 3 min @ 95 °C | 2 min @ 65 °C + 6 min @ 95 °C | 3 min @ 65 °C + 7 min @ 95 °C |
| Exposure energy | 125 mJ/cm2 | 155 mJ/cm2 | 170 mJ/cm2 |
| Post-exposure bake | 4 min @ 95 °C | 1 min @ 65 °C + 6 min @ 95 °C | 2 min @ 65 °C + 7 min @ 95 °C |
| Development | 2 min 30 s | 5 min | 6 min |
| Hard bake (optional) | 3-5 min @ 200 °C | 3-5 min @ 200 °C | 3-5 min @ 200 °C |
Table 2: Photolithography steps performed to build a device containing a central chamber of 10 μm in height. Heights of the intermediate chambers: 20, 50 and 90 µm.
| Step | Mask 1:12 µm layer | Mask 2:32 µm layer | Mask 3:40 µm layer |
| SU-8 type | 2015 | 2025 | 2050 |
| Spincoating | 30 s @ 3250 rpm | 30 s @ 2500 rpm | 30 s @ 3250 rpm |
| Soft bake | 3 min @ 95 °C | 2 min @ 65 °C + 5 min @ 95 °C | 3 min @ 65 °C + 7 min @ 95 °C |
| Exposure time | 140 mJ/cm2 | 157 mJ/cm2 | 170 mJ/cm2 |
| Post-exposure bake | 4 min @ 95 °C | 1 min @ 65 °C + 5 min @ 95 °C | 2 min @ 65 °C + 7 min @ 95 °C |
| Development | 3 min | 5 min | 6 min |
| Hard bake (optional) | 3-5 min @ 200 °C | 3-5 min @ 200 °C | 3-5 min @ 200 °C |
Table 3: Photolithography steps performed to build a device containing a central chamber of 3 μm in height. Heights of the intermediate chambers: 18, 50 and 90 µm.
Supplementary data 1: masks_neuron_volume_chips.tiff. Schematic view of the masks used to fabricate the PDMS device (DRIE mask and masks 1-3). Please click here to download this file.
Supplementary data 2: file "masks_neuron_volume_chips.dxf". Electronic files allowing to fabricate the DRIE mask and masks 1-3. Please click here to download this file.
Supplementary data 3: "Mask_Photoresist-stripes.dxf". Electronic files allowing to fabricate the mask used for the photolithography of photoresist stripes. Please click here to download this file.
Supplementary data 4: file conversion_mattotiff.m Please click here to download this file.
Supplementary data 5: file importfilevol.m Please click here to download this file.
