Simultaneous imaging of chemotaxis of multiple HL60 cells using cell mobility analysis device
Based on the principle of microfluidics16, the manufacturer has provided simulated profiles of gradients: a gradient is generated within 1 min, stabilized within 5 min, and maintained over 2 hr. The highly predictable profiles of the stable gradients generated by microfluidics allow multiple chemotaxis assays to be carried out simultaneously. In the present study, we observed three simultaneous chemotaxis assays (Figure 2A and Movie 1). We found that HL60 cells started chemotaxing immediately after the chemoattractant was injected into the well of the chemoattractant, and kept chemotaxing in a straight path for the following 60 min, consistent with the simulation results for gradient stability. Tracing the travel path and morphology of the cells allows quantitative measurements and subsequent comparison of the chemotaxis behaviors using a chemotaxis index that includes total path length, speed, directionality, and roundness of the cells (Figure 2B). Total path length is the sum of the lengths of the line segments connecting the centroids of the path. Speed is obtained by dividing the total path length by the time. Directionality is measured upward and is defined as: (Y coordinate of the end of the path minus Y coordinate of the beginning) divided by total path length. This gives 1.0 for an object moving directly upward. The roundness of the cell is a measure (in percent) of how efficiently a given amount of perimeter encloses area. A circle has the largest area for any given perimeter and has a roundness parameter of 100%. A straight line encloses no area and has a roundness parameter of 0%. We show the quantitatively measured chemotaxis behavior as described by the selected chemotaxis parameters (Figure 2C).
Monitoring PKD subcellular localization in HL60 cells under a spatiotemporally visible and controllable fMLP stimulus
It is a great technical advance to apply fluorescently labeled and controllable chemoattractant stimulation to an experimental system. Historically, we have applied either homogeneous (also called uniform) stimulation or gradient stimulation to observe cell response and behaviors. However, "blind" stimulation not only provides no spatiotemporal information on how the stimulus reaches the cells, but also casts doubt on any "abnormal" observations of cell response to stimulation, simply because we do not see the stimulus. We have previously shown that fluorescent dye (Alexa594) can be applied with chemoattractant to establish a linear relationship between chemoattractant concentration and monitored fluorescent dye intensity15. With an acquisition configuration of green fluorescent protein (GFP), a red emission of fluorescent dye (Alexa594), and transmitted light, we are able to monitor the adhering cells, the application of the stimulus, and the cell response to the stimulus (Figure 3A). Protein kinase D is a family of serine/threonine kinases that play essential roles in directed cell migration9,17. In response to uniformly applied fMLP (red) stimulation, HL60 cells mediate a robust membrane translocation of GFP-tagged protein kinase D1 (green) (Figure 3B and Movie 2). In an fMLP gradient (red) (Figure 4A), HL60 cells actively recruit PKD1 to the leading edge (Figure 4B and Movie 3). A close comparison of the subcellular localization of GFP in the protrusion of the leading edge indicates that PKD1 localizes at the rear of the leading edge (Figure 4C).
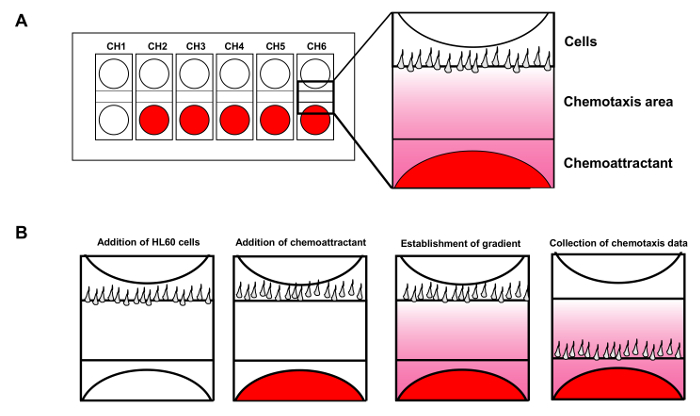
Figure 1. Cell mobility analysis device allows up to 6 simultaneous chemotaxis assays. (A) Scheme shows the design of a cell mobility analysis device chip for simultaneous monitoring of 6 independent chemotaxis assays. Red shows chemoattractant added to the wells. (B) Introduction of HL60 cells to the wells of cells while the chemoattractant diffuses to establish a steady fMLP gradient. Please click here to view a larger version of this figure.
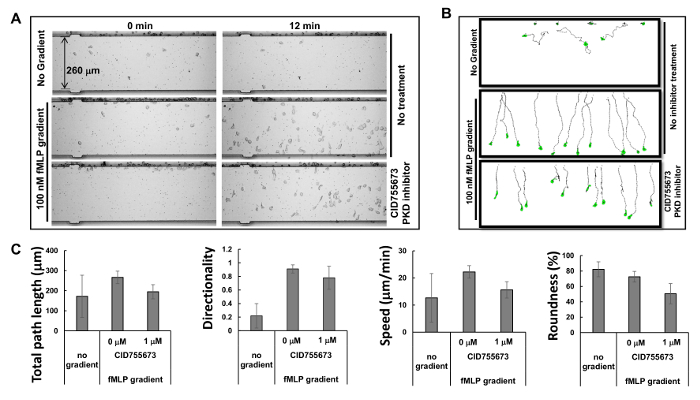
Figure 2. Simultaneous monitoring of multiple chemotaxis assays with HL60 cells. (A) Montage shows images of cell mobility analysis device chemotaxis assay to examine the inhibitory effects of PKD-specific inhibitors on chemotaxis at times of 0 and 12 min after gradient application. Chemotactic HL60 cells were pre-treated with PKD inhibitor 1 μM CID755673 for 30 min. HL60 cells with or without the treatment of PKD inhibitor were allowed to chemotax in either RPMI1640 starving medium or 100 nM fMLP gradients for 12 min. (B) Scheme shows the travel path length and morphology of traced HL60 cells. (C) Quantification of chemotaxis as total path length, speed, directionality, and roundness. Mean ± SD is shown; n = 10, 12, or 11 for no gradient, fMLP gradient without CID755673 treatment, and fMLP treatment with CID755673 treatment, respectively. Please click here to view a larger version of this figure.
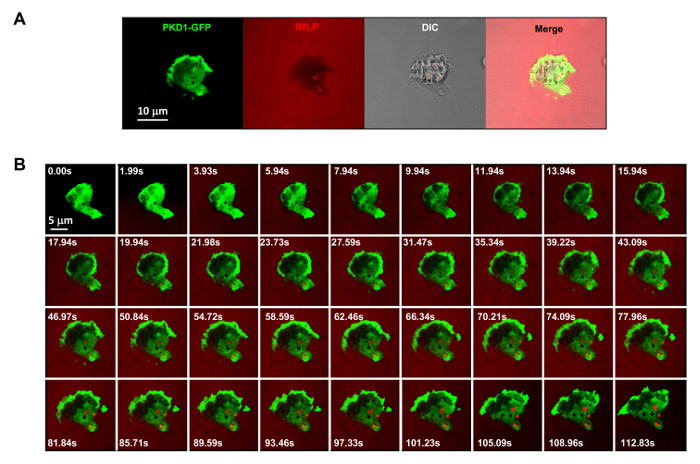
Figure 3. GPCR-mediated robust membrane translocation of PKD1 in response to uniformly applied fMLP stimuli. (A) Multichannel monitoring of PKD1-GFP (green), chemoattractant (1 μM fMLP mixed with 0.1 μg/ml fluorescent dye Alexa 594, red), and DIC (differential interference contrast) to identify the adhering HL60 cells in a well of a 4well chamber coated with 0.2% gelatin in RPMI 1640 medium. Scale bar = 10 μm. (B) Montage shows that uniformly applied fMLP (red) induces robust membrane translocation of PKD1-GFP (green). Please click here to view a larger version of this figure.
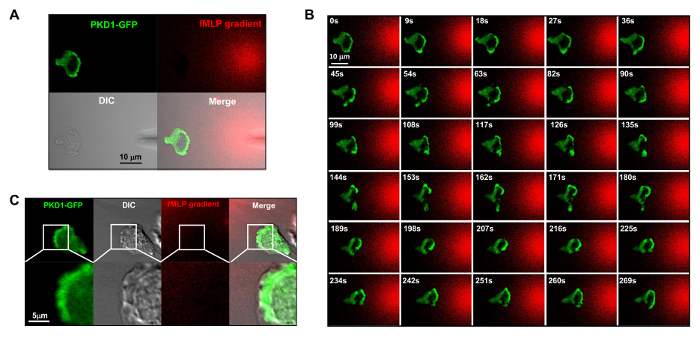
Figure 4. Leading edge localization of PKD1 in chemotaxing HL60 cells. (A) Channel mode acquisition configuration facilitates the visualization of the fMLP gradient and the spatiotemporal dynamics of PKD1. In A–C, HL60 cells transiently expressed GFP-tagged PKD1; to visualize the fMLP gradient generated from a micropipette (DIC), 100 nM fMLP (Red) was mixed with 0.1 μg/ml fluorescent dye Alexa 594. (B) Enriched localization of PKD1 at the leading edge of the chemotaxing cell. Scale bar = 10 μm. (C) Merged images show that PKD1 localizes at the rear of the leading edge in HL60 cells. Green shows PKD1 cellular localization, and the DIC image shows the protruding area of the leading edge. Scale bar = 5 μm. Please click here to view a larger version of this figure.
