Methods to Study Mrp4-containing Macromolecular Complexes in the Regulation of Fibroblast Migration
Summary
MRP4 regulates various cyclic nucleotide-dependent signaling events including a recently elucidated role in cell migration. We describe a direct, but multifaceted approach to unravel the downstream molecular targets of MRP4 resulting in identification of a unique MRP4 interactome that plays key roles in the fine-tuned regulation of fibroblast migration.
Abstract
Multidrug resistance protein 4 (MRP4) is a member of the ATP-binding cassette family of membrane transporters and is an endogenous efflux transporter of cyclic nucleotides. By modulating intracellular cyclic nucleotide concentration, MRP4 can regulate multiple cyclic nucleotide-dependent cellular events including cell migration. Previously, we demonstrated that in the absence of MRP4, fibroblast cells contain higher levels of intracellular cyclic nucleotides and can migrate faster. To understand the underlying mechanisms of this finding, we adopted a direct yet multifaceted approach. First, we isolated potential interacting protein complexes of MRP4 from a MRP4 over-expression cell system using immunoprecipitation followed by mass-spectrometry. After identifying unique proteins in the MRP4 interactome, we utilized Ingenuity Pathway Analysis (IPA) to explore the role of these protein-protein interactions in the context of signal transduction. We elucidated the potential role of the MRP4 protein complex in cell migration and identified F-actin as a major mediator of the effect of MRP4 on cell migration. This study also emphasized the role of cAMP and cGMP as key players in the migratory phenomena. Using high-content microscopy, we performed cell-migration assays and observed that the effect of MRP4 on fibroblast migration is completely abolished by disruption of the actin cytoskeleton or inhibition of cAMP-dependent kinase A (PKA). To visualize signaling modulations in a migrating cell in real time, we utilized a FRET-based sensor for measuring PKA activity and found, the presence of more polarized PKA activity near the leading edge of migrating Mrp4-/- fibroblast, compared to Mrp4+/+fibroblasts. This in turn increased cortical actin formation and augmented the process of migration. Our approach enables identification of the proteins acting downstream to MRP4 and provides us with an overview of the mechanism involved in MRP4-dependent regulation of fibroblast migration.
Introduction
Cell migration is a complicated multi-step process. Studies have shown that during migration cells are polarized into leading and trailing edges. By adhering to the extracellular matrix, the leading edge provides the traction necessary for the cell body to move forward. Finally, the trailing edge releases rear attachments and completes the migration cycle1,2.
Cell polarization for efficient cell migration is regulated by spatial segregation of intracellular signaling. Cellular second messengers, such as cAMP, mediate the compartmentalization of the signaling events required for fine-tuned directional cell migration3,4. Preferential accumulations of cAMP and cAMP-dependent kinase PKA activity at the leading edge play key roles in directional cell migration5,6. By phosphorylating small GTPases such as Ras-related C3 botulinum toxin substrate (Rac) and cell division control protein 42 homolog or Cdc42, PKA activates actin-related protein 2/3 (Arp 2/3) at the leading edge and induces the formation of lamellipodia7-9. PKA also phosphorylates an anti-capping agent, vasodilator stimulated phosphoprotein (VASP), thereby regulates the oscillatory cycles of membrane extension and retraction10,11.
In cells, cAMP levels are regulated by three major processes: i) synthesis by adenylate cyclase, ii) degradation by phosphodiesterases, and iii) transportation by membrane-bound efflux transporters3. Multidrug resistance protein 4 (MRP4), a member of ATP-binding cassette (ABC) family of membrane transporters, functions as an endogenous efflux transporter of cyclic nucleotides. Therefore, MRP4 can regulate intracellular cAMP levels and cAMP-dependent cellular signaling11-13. We have previously shown that in Mrp4-/-, fibroblasts contain relatively higher levels of cyclic nucleotides and migrate faster compared to Mrp4+/+ fibroblasts14. We also reported a biphasic effect of cyclic nucleotides on fibroblast migration. Based on previous studies and our finding that Mrp4-/- fibroblasts contain more polarized cAMP during the course of migration, we hypothesized that this MRP4-mediated regulation of fibroblast migration is cAMP dependent. In order to understand the downstream mechanism, we took a direct yet multifaceted approach.
To identify the proteins associated and in interplay with MRP4, we immunoprecipitated MRP4-containing macromolecular complexes from HEK293 cells that over express MRP4. Using mass-spectrometry, we identified multiple MRP4-interacting proteins and analyzed their interconnectivity using Ingenuity Pathway Analysis (IPA). IPA is a useful tool to analyze protein-protein interactions (both structural and functional) and explore their contributions in particular physiological and pathological events based on the literature and experimental evidences15,16. IPA indicated that F-actin is a major downstream target of MRP4 in the context of cell migration where cAMP and cGMP are the key signaling molecules17. These data were further confirmed by high-content microscopy. High-content microscopy can capture and analyze cell behaviors such as cell migration in a more convenient, accurate and high-throughput manner18. The high-content microscopy data demonstrated that the effect of MRP4 on fibroblast migration is completely abolished upon disruption of the actin cytoskeleton or inhibition of PKA17.
Additionally, we used a Förster resonance energy transfer (FRET)-based PKA sensor to monitor the PKA dynamics in migrating cells in real time. FRET-based kinase sensors usually consist of specific phosphorylation substrate peptides flanked by CFP and YFP fluorophores19-21. pmAKAR3 is an improved and membrane targeted FRET-based PKA sensor that contains forkhead-associated domain 1 (FHA1) and the PKA substrate sequence LRRATLVD5,22. Phosphorylation of pmAKAR3 by the PKA catalytic subunit increases FRET signal between CFP and YFP19. Insertion of a lipid modification domain into the sensor targets it to the plasma membrane for monitoring PKA dynamics, specifically at the membrane compartment23.
Using pmAKAR3, we demonstrated that the leading edge of migrating Mrp4-/- fibroblasts exhibited more polarized PKA activity than Mrp4+/+ fibroblasts, which in turn increased the cortical actin formation at the cell's leading edge17. Together, these events resulted in better cellular polarization and faster directional cell migration in the absence of MRP4. Our specific and direct approach identified key downstream targets for MRP4 and provides an important, but as of yet unexplored mechanism for MRP4-dependent regulation of fibroblast migration.
Protocol
1. Ingenuity Pathway Analysis
- Uploading Protein-interactome Dataset
- Insert the proteins/genes of interest in a spreadsheet with their unique gene identifiers (preferably gene symbols and gene identifier numbers as obtained with mass spectrometric data).
- Assign one column in the spreadsheet for Gene identifier number and one column for observational value (e.g., Fold-change or p-value). Select the 'contains column header' option to view the column headings.
- Upload the dataset on the IPA by clicking on the upload dataset tab and selecting the spreadsheet mentioned above. Choose the flexible format tab, and select the appropriate category of gene identifier.
Note: A flexible format of the dataset overcomes the formatting specifications for uploading. - After the dataset appears, select ID and observation columns. Ensure that all of the proteins (MRP4 interactome as identified by mass-spectrometry) are mapped by checking on the dataset summary tab. The dataset is now ready for the desired analysis.
- Data Analysis
Note: Described are the partial uses of IPA features that were utilized for this study.- After uploading MRP4 interactome with their gene names as the unique identifiers, click on new and choose core analysis in the top left menu of IPA software.
- Set the expression cutoffs value for intensity in accordance with the experiment type (Intensity value of 100 is recommended for analysis).
- Calculate the fold-change in expression while preparing the dataset if control and experimental conditions are involved and include it as part of the dataset file if a comparison analysis needs to be performed (A cut-off value of 1.5 fold change is usually recommended for analysis).
Note: After completion of the core analysis, multiple sections of analyses can be obtained. The first tab shows the summary of the total analyses and suggests top canonical pathways, upstream regulators, molecular and cellular functions, and networks among others; all calculated based on the confidence level (p value) and arranged in ascending order of p value. - Open the major canonical pathways (using the open pathway option) as large molecular networks that pictorially show cross talking, overlapping, and affected signaling pathways.
Look at the list of the canonical pathways that are majorly involved based on the p-value (p <0.05 is considered as significant).
Note: The significance values for the canonical pathways are calculated by Fisher's exact test right-tailed. The significance indicates the probability of association of molecules from the dataset with the canonical pathway by random chance alone. - Confirm the input proteins (MRP4 interactome as identified by mass-spectrometry) among the list of proteins involved in each pathway.
Note: Using this option, it was observed that the cellular actin cytoskeleton signaling network is the major affected pathway and how the input proteins fit into it (Figure 1). This approach helps identify which molecular networks would be closely associated and affected by the test proteins. - Use the network tab in the analyses to identify the top diseases and functions that are potentially linked to a particular protein network based on the number of focus molecules.
Note: A score value is given based on the known proteins that are determined as part of a network e.g., cellular assembly and organization network, through literature knowledge and experimental evidences and focus molecules that are identified from the dataset and become part of the network. Henceforth, the network gets filtered in the first place based on the number of focus molecules.
2. High-content Microscopy
- Preparation of Cells
Note: All cell culture work was done under the horizontal-flow cell culture hood.- Use 96-well microplates for the wound-healing assay.
- Coat the microplate with 1% fibronectin solution (reconstituted in PBS), and keep the plate in a standard CO2 incubator at 37 °C for 2 hr.
- Remove Mrp4-/- and Mrp4+/+ mouse embryonic fibroblast (MEFs) grown in 25 cm² cell culture flasks from the incubator, wash 1 time with PBS, and trypsinize the cells for 5 min using 1 ml of 4x trypsin-EDTA solution.
- Wash the cells with complete medium (DMEM containing 10% FBS and 1% penicillin/streptomycin) and resuspend them in 5 ml of complete medium.
- Aspirate the fibronectin solution from the wells. Count cells using a hemocytometer and seed 30,000 cells (cell number needs to be optimized based on cell type) into each well.
- Grow the cells to 100% confluence in a standard CO2 incubator at 37 °C for 24 hr.
- Creating Wound
- Ensure all the wells are filled with solution. Fill unused wells with PBS to prevent damage to the wound making tool (e.g., WoundMaker).
- After confirming 100% cell confluence, place the 96-well plate inside the wound making tool on the metal base plate and press the 96-well pin block down.
- Wash the cells three-times with PBS to remove any dislodged cells and add 100 µl of complete medium.
- Add the test compounds – H-89 (50 µM; 1:1,000 dilution with complete media using 50 mM stock in DMSO) or Latrunculin B (1 µM; 1:1,000 dilution with complete media using 1 mM stock in DMSO) at this stage.
- Place the assay plate inside the reader at 37 °C and monitor migration over the experimental period.
- Monitoring Cell Migration
Note: Cell migration was monitored using the microscope and software provided with high-content microscopy system. The use of 96-well microplates ensures that the wounds are automatically monitored by the microscope and registered by the software.- Set the software to scan the assay plate every hour for migration assays using the schedule scan tab . Select a start time at least 15 min after placing the assay plate inside the reader to allow equilibration prior to imaging.
- Select the tray and choose vessel type (96-well microplate) and experiment type (Scratch Wound).
- Select 10X objective and choose phase-contrast imaging parameters. Select the wells that need to be scanned using the edit scan pattern option.
- Specify the treatments groups and any replicates by selecting the properties tab and setting up a plate map. Scanning can be stopped at any time based on the experimental condition.
- Data Analysis
- After the scanning is done, select the vessel view tab for the assay plate. Go to the analysis job utilities and select launch new analysis job.
- Set scratch wound as job type and select the time range for the analysis (0 time point to 24 hr time point).
- Select the wells to be chosen for analysis by a single click on the well and click 'OK' to initiate the analysis. Once the analysis is done, graphically visualize relative wound density (RWD) data for each well at each time point using export to the graph option.
- View the phase-contrast image set of each well corresponding to different time-points using the view image tab. Select a specific well by clicking on the plate map, select a specific time point from the time range tab and click on the view image tab.
3. Förster Resonance Energy Transfer (FRET)
- Preparation of Cells
- Coat 35 mm glass-bottomed dishes with 100 µl of 1% fibronectin solution (as described in section 2.1) and keep them in a standard CO2 incubator at 37 °C for 2 hr.
- Aspirate the fibronectin solution from the plates and seed equal numbers of HEK293 cells (10,000 cells/plate) in complete medium.
- Grow the cells to 60-70% confluence in the fibronectin coated glass-bottomed dishes in a standard CO2 incubator at 37 °C for 24 hr.
- Transfection
- Perform all transient transfections using a commercial transfection reagent according to manufacturer's instructions.
- Aliquot 250 µl of complete medium into two separate 1.5 ml sterile centrifuge tubes for each 35 mm dish.
- Add 2 µg of DNA (pmAKAR3 or pmAKAR3-TA) in one tube and 5 µl (2.5 fold the DNA concentration) of transfection reagent in another tube.
- Incubate the tubes for 20 min at room temperature and then transfer the diluted DNA into the transfection reagent-containing tube.
- Mix thoroughly and incubate at 37 °C for another 30 min.
- Aspirate the medium off the cells and wash them once with PBS. Add 1 ml of antibiotic-free DMEM F-12 medium containing 10% FBS to the cells.
- Add 507 µl of DNA-lipid conjugate with media to the cells in each plate, mix gently, and keep in the CO2 incubator at 37 °C for 48 hr. Use 2 µg of DNA (1 µg/µl stock) and 5 µl lipid for 10,000-50,000 cells in each plate.
- Live-cell Imaging
- After 48 hr of transfection, remove growth medium and wash the cells 2 times with Hank's Balanced Salt Solution (HBSS, pre-warmed at 37 °C). Add a final volume of 1.9 ml HBSS to the washed cells and mount them on an inverted wide-field microscope system for FRET imaging inside a custom-made 37 °C maintained chamber.
Note: In this system, the excitation light is provided by a 300 W Xenon lamp attenuated with a Neutral Density filter with 50% light transmission. - Use 60X objective. Manually focus on the cells and set an optimal field of view using the microscope. Activate the selected field of view on the computer screen using the Focus "F" option on the software. For performing FRET measurements, select the proper filter set (CFP/YFP filter set with a 430/25 nm excitation filter, a double dichroic beam splitter, and two emission filters (470/30 nm for CFP and 535/30 nm for FRET) manually.
- Visualize fluorescence by first checking on the channel option CFP/YFP/FRET followed by clicking open fluor tab. Improve the signal intensity by using the intensification tool in the camera tab.
- Select cells expressing pmAKAR3 or pmAKAR3-TA avoiding saturation of the signal intensity. Use the capture window to initiate FRET measurement.
- Select the time-lapse option and setup a time-scan for 30 min with 30 sec interval. Check the FRET option and set the exposure time at 100 msec. Enter the image label and press start to initiate the measurement.
- After five time points and establishment of a baseline, add 25 µM of forskolin (5 µl of 10 µM stock in ethanol, diluted with 100 µl of HBSS) to the cells without disturbing the plate and monitor the cells for a total of 30 min.
- After 48 hr of transfection, remove growth medium and wash the cells 2 times with Hank's Balanced Salt Solution (HBSS, pre-warmed at 37 °C). Add a final volume of 1.9 ml HBSS to the washed cells and mount them on an inverted wide-field microscope system for FRET imaging inside a custom-made 37 °C maintained chamber.
- Data Analysis
Note: Use ratiometric calculation module for data analysis.- Click on the select tool on the left-hand side of the image window and choose a solid rectangle from the drop down menu. Drag the rectangle on the image field in a cell free area and right click on it to set it as background for the purpose of background subtraction.
- Go to the mask and use 'create a new mask' option. Go to the select tool and choose a pen from the drop down menu to manually draw a mask around the cell to select it for a measurement. Select at least 4-6 cells per condition.
- Select the Mask tab and perform mask statistics.
- Check 'Entire mask', expand cross-channel option and select donor normalized FRET (N-FRET) (FRET/CFP). The N-FRET value is representative of PKA activity at the plasma membrane.
- Add time-stamps, look-up table and scale bar using the annotations.
Representative Results
To study the effect of MRP4 on fibroblast migration, we used a wound-healing assay utilizing high-content microscopy14. Precise wounds were made on confluent monolayers of MEFs isolated from either Mrp4-/- or Mrp4+/+ mice, and images were taken at 1 hr intervals for 24 hr. We observed a higher migration rate for Mrp4-/- MEFs compared to Mrp4+/+ MEFs (Figure 2). The wounds were completely healed in less than 15 hr for the Mrp4-/- MEFs, whereas the Mrp4+/+ MEFs required almost 20 hr to cover the wounds.
Polarized accumulation of PKA activity at the leading edge of migrating cells is a key early event for directional migration. PKA activity can be monitored in real time using the pmAKAR3 FRET-based sensor for PKA5,17. To check the specificity of pmAKAR3 for PKA activity, we treated HEK293 cells overexpressing pmAKAR3 or the point mutant pmAKAR3-TA, which contains a threonine-to-alanine mutation at the substrate region for PKA and therefore is irresponsive to PKA phosphorylation with 25 µM forskolin, a cAMP-inducing agent14. We found an increase in FRET signal in cells overexpressing pmAKAR3, but cells overexpressing pmAKAR3-TA remained unchanged (Figure 3). The basal FRET levels were also higher for cells expressing pmAKAR3 compared to the cells expressing pmAKAR3-TA. These data indicated that pmAKAR3 is very specific for PKA activity.
In summary, the methods described in the protocol section are useful tools to study the molecular mechanism associated with a specific cellular event.
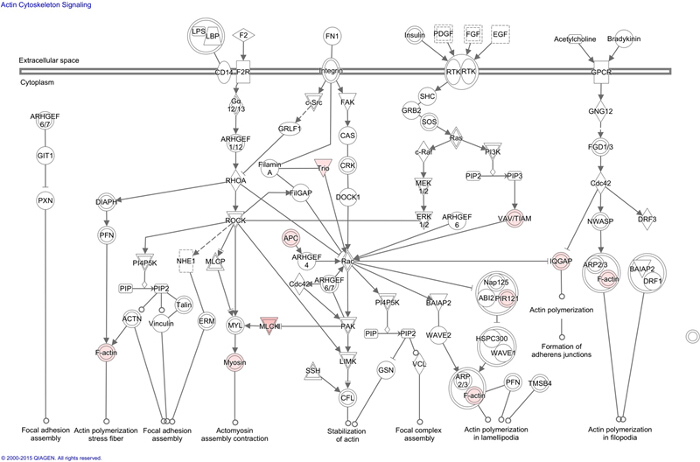
Figure 1: Ingenuity Pathway Analysis (IPA) of MRP4 Interactome. Using IPA actin cytoskeleton pathway was identified as one of the major canonical pathways affected by MRP4 interactome. Presented actin signaling network indicates connected proteins (white) and their cross communication with the proteins identified in the MRP4 interactome (pink) inferred from literature and experimental evidences. Please click here to view a larger version of this figure.

Figure 2: Wound-healing Assay using High Content Microscopy. Mrp4+/+and Mrp4-/- mouse embryonic fibroblasts (MEFs) were grown on fibronectin-coated 96-well dishes, and wounds in the monolayers were made precisely using the 96-pin wound-maker. Representative images at different time points are shown with 10X magnification. Please click here to view a larger version of this figure.
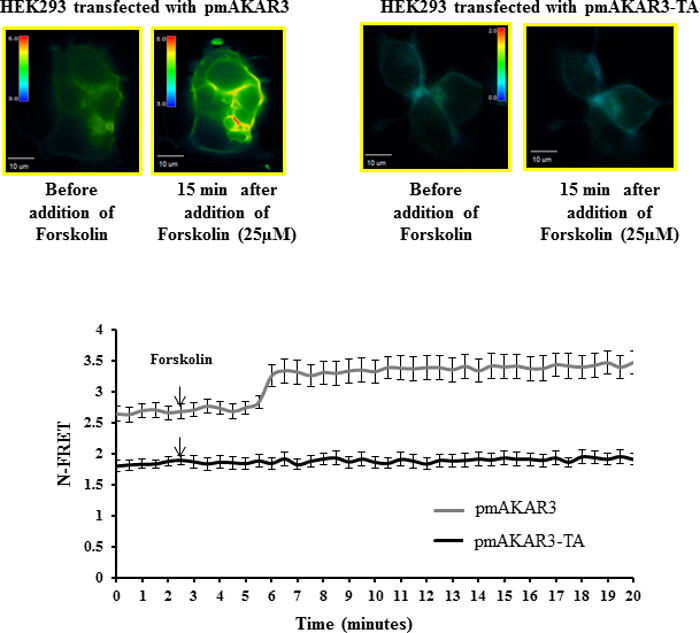
Figure 3: Förster Resonance Energy Transfer (FRET)-based Measurement of PKA Activity using pmAKAR3 Sensors. Representative pseudo-color images of N-FRET with 60X magnification for HEK293 cells transfected with pmAKAR3 sensor and pmAKAR3-TA sensor before and after forskolin treatment are shown (top panels). Images in each panel were captured from the same field of view. Color bar indicates the magnitude of the N-FRET. The line graph (bottom panel) represents the change in N-FRET levels after treatment with forskolin. Data represent the average of at least three independent experiments (Mean ± SEM; n = 3). Please click here to view a larger version of this figure.
Discussion
Cell migration is an intricate process that plays indispensable roles in many important physiological events including wound healing1,2. Aberrant cell migrations may cause catastrophic events, such as tumor metastasis and angiogenesis24,25. Therefore, fine-tuned regulation of cell migration is required to maintain normal body function.
Using high-content microscopy18, we demonstrated that MRP4-deficient MEFs migrate faster compared to wild-type fibroblasts14. In contrast to the conventional scratch wound assay, the microscopy here conducts the wound healing assay in an automated convenient, consistent and high-throughput manner. The software analyzes the cell migration rate based on three separate metrics: i) change in wound width, ii) change in cell confluence within the wound region, and iii) relative wound density (RWD). RWD is a self-normalized metric that measures the spatial cell density inside the wound area relative to the spatial cell density outside of the wound area. Therefore, it is not affected by changes in cell density due to cell proliferation and provides very specific information regarding cell migration which is otherwise difficult to be obtained by the conventional scratch wound assay26. Initially, at the 0 time point, the RWD will be 0% and upon complete wound healing, the RWD will be 100%. All of these metrics are calculated by custom-made software with inbuilt algorithms and the migration information is automatically generated for every time point. The assay is easy to perform but proper washing (70% ethanol) and handling (inside the hood) of the wound-maker is important to prevent contamination. Our data suggested that the RWD kinetics for Mrp4-/- MEFs are significantly higher compared to Mrp4+/+ MEFs.
During migration, cells polarize into leading and trailing edges that ultimately pull the cell body toward the direction of migration1,27. Distinct and segregated signaling events at different regions of a moving cell ensure the polarization process. Polarized accumulation of cAMP and subsequent activation of PKA at the leading edge is a key early step in directional cell migration5. Since MRP4 has very high affinity for cAMP (Km = 45 µM)12, we hypothesize that the effect of MRP4 on cell migration is cAMP-dependent. To identify the proteins acting downstream of MRP4 and simultaneously interacting with MRP4, we characterized MRP4-containing macromolecular complexes by mass-spectrometry. The MRP4 interactome was subjected to multiple analyses including generation of protein networks, path maps, and functional integration to the canonical pathways of the cellular signaling and their respective pictorial representation through the use of IPA. In general, IPA allows scientific users to recognize the molecular and physiological contexts of their genes and proteins of interest. However the analysis is completely based on the literature and experimental evidences. Novel interactions cannot be suggested by IPA. But it can identify which network, the proteins of interest, can potentially form. Additionally users can identify the top diseases and functions that are potentially linked to a particular protein network based on the confidence level (P value). Of interest to our study, the actin cytoskeleton pathway was a major affected pathway with a P value of 6.75 x 10-4. This comprehensive approach also revealed that F-actin is a major protein target for MRP4 and cAMP is the key mediator17. Based on these data, we further studied the underlying molecular mechanisms.
To understand the effect of MRP4 on cAMP dynamics and PKA activity during the course of cell migration, we used FRET-based live-cell imaging techniques. Using FRET-based sensors for cAMP and PKA activity, we confirmed higher cAMP accumulation and higher PKA activity at the leading edge of migrating and polarized fibroblasts22,28. We further demonstrated that in the absence of MRP4, MEFs have more polarized cAMP and PKA activity, which in turn facilitates cortical actin formation and cell migration. The high-content microscopy-based wound-healing assay showed that the effect of MRP4 on cell migration is completely abolished by PKA inhibition or actin disruption, which indicates a direct role of PKA and actin as downstream targets17. Unlike conventional cell population-based assays such as ELISA, use of a FRET-based sensor allows us to identify the downstream effector kinases that regulate various signaling processes and detect the correlation between cyclic nucleotide dynamics and their corresponding kinase activity in real time and space. Additionally it can discriminate intracellular and intercellular heterogeneity during the signaling events. For example it can detect the difference in PKA dynamics in the cells at the wound edge compared to the cells inside the monolayer and away from the wound edge5, whereas ELISA based assays can only detect total intracellular cyclic nucleotide or PKA level in a sample14,17. However the transfection efficacy of particular cell types can be a limiting factor for conducting FRET based assays but the highly efficient transfection reagents can overcome this problem.
Together, our results indicate that in addition to conferring drug resistance, MRP4 also plays important physiological roles by modulating intracellular cAMP dynamics. Using three unique approaches, i) high-content microscopy18, ii) IPA15, and iii) FRET5,28, we have begun to unravel the previously undefined role of MRP4 in cell migration. In general, these useful scientific techniques will allow us to identify new downstream targets of any protein of interest and explore novel molecular mechanisms involved in particular pathological or physiological cell responses. Where IPA provides useful information regarding downstream effectors of the protein of interest and potential regulatory networks; FRET-based live imaging can monitor compartmentalized signaling in real time. High-content microscopy is a convenient high-throughput screening tool to monitor and analyze physiological events, such as cell migration and cell proliferation, over a period of time as a final readout.
Divulgazioni
The authors have nothing to disclose.
Acknowledgements
This work was supported by National Institutes of Health grants R01-DK080834 and R01-DK093045. We thank J. Denise Wetzel, CCHMC Medical Writer, for editing of the manuscript.
Materials
| Lipofectamine 2000 | Invitrogen(Carlsbad, CA) | 11668-027 | |
| DMEM | Invitrogen (Carlsbad, CA) | 11965-092 | |
| IncuCyte Zoom | Essen BioScience | ||
| 96-well IncuCyte Image-Lock microplates | Essen BioScience | 4493 | |
| Latrunculin B | Sigma-Aldrich (St. Louis, MO). | L5288 | Stock in DMSO |
| H-89 | Enzo Life Sciences (Farmingdale, NY) | BML-EI196 | Stock in DMSO |
| 35-mm glass-bottomed dishes | (MatTek Corporation; Ashland, MA) | P35G-1.5-20-C | |
| Fibronectin | Sigma-Aldrich (St. Louis, MO). | F1141 | |
| Opti-MEM Reduced Serum Media | Invitrogen (Carlsbad, CA) | 31985-088 | |
| FRET microscopy system | Olympus inverted microscope (IX51) | ||
| CCD camera | Hamamatsu, Japan | ORCA285 | |
| SlideBook software 5.5 | Intelligent Imaging Innovation ( Denver, CO) | ||
| Ingenuity Pathway Analysis software | IPA, QIAGEN Redwood City, | ||
| Forskolin | Tocris (Ellisville, MO). | 1099 | Stock in100% EtOH |
| DMEM F-12 | Invitrogen (Carlsbad, CA) | 11330-057 | |
| HBSS | Invitrogen (Carlsbad, CA) | 14025-134 | |
| Excel | Microsoft | ||
| PBS | Invitrogen(Carlsbad, CA) | 10010-023 | |
| Trypsin/EDTA Solution (TE) | Invitrogen(Carlsbad, CA) | R-001-100 | |
| Penicillin-Streptomycin | Invitrogen(Carlsbad, CA) | 15140-122 |
Riferimenti
- Ridley, A. J., et al. Cell migration: integrating signals from front to back. Science. 302 (5651), 1704-1709 (2003).
- Lauffenburger, D. A., Horwitz, A. F. Cell migration: a physically integrated molecular process. Cell. 84 (3), 359-369 (1996).
- Arora, K., et al. Compartmentalization of cyclic nucleotide signaling: a question of when, where, and why?. Pflugers Arch. 465 (10), 1397-1407 (2013).
- Howe, A. K., Baldor, L. C., Hogan, B. P. Spatial regulation of the cAMP-dependent protein kinase during chemotactic cell migration. Proc Natl Acad Sci U S A. 102 (40), 14320-14325 (2005).
- Lim, C. J., et al. Integrin-mediated protein kinase A activation at the leading edge of migrating cells. Mol Biol Cell. 19 (11), 4930-4941 (2008).
- Paulucci-Holthauzen, A. A., et al. Spatial distribution of protein kinase A activity during cell migration is mediated by A-kinase anchoring protein AKAP Lbc. J Biol Chem. 284 (9), 5956-5967 (2009).
- Weaver, A. M., Young, M. E., Lee, W. L., Cooper, J. A. Integration of signals to the Arp2/3 complex. Curr Opin Cell Biol. 15 (1), 23-30 (2003).
- Le Clainche, C., Carlier, M. F. Regulation of actin assembly associated with protrusion and adhesion in cell migration. Physiol Rev. 88 (2), 489-513 (2008).
- Raftopoulou, M., Hall, A. Cell migration: Rho GTPases lead the way. Dev Biol. 265 (1), 23-32 (2004).
- Krause, M., Dent, E. W., Bear, J. E., Loureiro, J. J., Gertler, F. B. Ena/VASP proteins: regulators of the actin cytoskeleton and cell migration. Annu Rev Cell Dev Biol. 19, 541-564 (2003).
- Hara, Y., et al. Inhibition of MRP4 prevents and reverses pulmonary hypertension in mice. J Clin Invest. 121 (7), (2011).
- Russel, F. G., Koenderink, J. B., Masereeuw, R. Multidrug resistance protein 4 (MRP4/ABCC4): a versatile efflux tra (7), 2888-289nsporter for drugs and signalling molecules. Trends Pharmacol Sci. 29 (4), 200-207 (2008).
- Cheepala, S., et al. Cyclic nucleotide compartmentalization: contributions of phosphodiesterases and ATP-binding cassette transporters. Annu Rev Pharmacol Toxicol. 53, 231-253 (2013).
- Sinha, C., et al. Multi-drug resistance protein 4 (MRP4)-mediated regulation of fibroblast cell migration reflects a dichotomous role of intracellular cyclic nucleotides. J Biol Chem. 288 (6), 3786-3794 (2013).
- Popovici, C., et al. Direct and heterologous approaches to identify the LET-756/FGF interactome. BMC Genomics. 7 (105), (2006).
- Soler-Lopez, M., Zanzoni, A., Lluis, R., Stelzl, U., Aloy, P. Interactome mapping suggests new mechanistic details underlying Alzheimer’s disease. Genome Res. 21 (3), 364-376 (2011).
- Sinha, C., et al. PKA and actin play critical roles as downstream effectors in MRP4-mediated regulation of fibroblast migration. Cell Signal. 27 (7), 1345-1355 (2015).
- Liu, L., Wang, Y. D., Wu, J., Cui, J., Chen, T. Carnitine palmitoyltransferase 1A (CPT1A): a transcriptional target of PAX3-FKHR and mediates PAX3-FKHR-dependent motility in alveolar rhabdomyosarcoma cells. BMC Cancer. 12 (154), (2012).
- Zhang, J., Ma, Y., Taylor, S. S., Tsien, R. Y. Genetically encoded reporters of protein kinase A activity reveal impact of substrate tethering. Proc Natl Acad Sci U S A. 98 (26), 14997-15002 (2001).
- Sinha, C., et al. Forster resonance energy transfer – an approach to visualize the spatiotemporal regulation of macromolecular complex formation and compartmentalized cell signaling. Biochim Biophys Acta. 1840 (10), 3067-3072 (2014).
- Sato, M., Ozawa, T., Inukai, K., Asano, T., Umezawa, Y. Fluorescent indicators for imaging protein phosphorylation in single living cells. Nat Biotechnol. 20 (3), 287-294 (2002).
- Allen, M. D., Zhang, J. Subcellular dynamics of protein kinase A activity visualized by FRET-based reporters. Biochem Biophys Res Commun. 348 (2), 716-721 (2006).
- Ananthanarayanan, B., Ni, Q., Zhang, J. Signal propagation from membrane messengers to nuclear effectors revealed by reporters of phosphoinositide dynamics and Akt activity. Proc Natl Acad Sci U S A. 102 (42), 15081-15086 (2005).
- Yamaguchi, H., Condeelis, J. Regulation of the actin cytoskeleton in cancer cell migration and invasion. Biochim Biophys Acta. 1773 (5), 642-652 (2007).
- Lamalice, L., Le Boeuf, F., Huot, J. Endothelial cell migration during angiogenesis. Circ Res. 100 (6), 782-794 (2007).
- Ghosh, M. C., Makena, P. S., Gorantla, V., Sinclair, S. E., Waters, C. M. CXCR4 regulates migration of lung alveolar epithelial cells through activation of Rac1 and matrix metalloproteinase-2. Am J Physiol Lung Cell Mol Physiol. 302 (9), L846-L856 (2012).
- Vicente-Manzanares, M., Webb, D. J., Horwitz, A. R. Cell migration at a glance. J Cell Sci. 118 (Pt 21), 4917-4919 (2005).
- Zaccolo, M., et al. A genetically encoded, fluorescent indicator for cyclic AMP in living cells. Nat Cell Biol. 2 (1), 25-29 (2000).
