Viral Metagenomes
CF sputum is exceptionally viscous and contains a high amount of mucin and free DNA (Figure 2A); the density gradient ultracentrifugation facilitates the elimination of host-derived DNA (Figure 2B). The results from a previous study9 showing eight viromes generated from the presented workflow are summarized here (Table 1). Seven samples (CF1-D, CF1-E, CF1-F, CF4-B, CF4-C, CF5-A, and CF5-B; Table 1) were processed as described in Section 2. The generated viromes contained little (0.02%-3.7%) human-derived sequences with only one exception (70%). CF4-A was omitted from the density gradient ultracentrifugation step (CF4-A) and the virome generated from this specific sample contained >97% human-derived sequences (Table 1). Figure 2 shows an example of the epifluorescence microscopy image of a typical CF sputum sample before (Figure 2A) and after (Figure 2C) density gradient ultracentrifugation. Clear viral-like particles (VLPs) were observed in the micrographs without large particles following the density gradient separation. After VLPs DNA extraction, bacterial contamination is often tested using 16S rDNA amplification prior to the sequencing of VLPs DNA.
Microbial Metagenomes
Seven sputum samples presented here were collected from a single CF patient across seven consecutive days. The patient started on oral antibiotic (Ciprofloxacin and Doxycycline) on Day 3 after the sputum was collected. The volume of each sputum sample collected from this patient was 15 ml throughout the 7 days; therefore, PBS was not added to the sample. The goal of this sampling event was to evaluate the protocols presented in this workflow by (i) evaluating the daily fluctuation of microbial community structure, and (ii) compare the microbial community structure and resolution between metagenomics and 16S rDNA sequencing. Therefore, total DNA and HL-DNA were extracted from each sample.
The HL-DNA concentration of each sputum sample following DNA extraction is presented in Table 2. The total yield of HL-DNA ranged from 210 ng to >5 μg. Illumina sequencing libraries were generated with a total starting material of 1 ng for each sample (Figure 3). The characteristics of the metagenomics data are presented in Table 2. All but one library yielded more than 1 million sequences and more than 85% high quality sequences were retained upon data preprocessing using the PRINSEQ29 software. All datasets were first preprocessed to remove duplicates and sequences of low quality (minimum quality score of 25), followed by further screening and removal of human-derived sequences using DeconSeq30. The amount of human-derived sequence contamination is highly dependent on the sample properties. Here, the total amount of human-derived sequences ranged from 14-46% (Table 2). The preprocessed sequences were then annotated using the Metaphlan31 pipeline as well as MG-RAST32 server.
In addition to metagenomes, 16S rDNA amplicon libraries were generated from both the total DNA and HL-DNA via primers targeting approximately 300 bp of the V1-V2 variable region in the 16S rRNA gene33,34. PCR products from individual samples were normalized and pooled for sequencing using the Illumina 500-cycle paired-end sequencing performed on the MiSeq platform. Paired-end 16S rDNA amplicon sequences were sorted by sample via barcodes using a python script and the paired reads were assembled using phrap35,36. Assembled sequence ends were trimmed until the average quality score was ≥20 using a 5 nt window. Potential chimeras were then removed using Uchime37 against a chimera-free subset of the SILVA38 reference sequences. Taxanomy was assigned to the high quality reads with SINA39 (version 1.2.11) using the 418,497 bacterial sequences from the SILVA38 database. Sequences with identical taxonomic assignments were clustered to produce Operational Taxonomic Units (OTUs). This process generated 1,655,278 sequences for 16 samples (average size: 103,455 sequences/sample; min: 72,603; max: 127,113). The median Goods coverage score, a measure of completeness of sequencing, was ≥ 99.9%. The software package Explicet40 (v2.9.4, www.explicet.org) was used for analysis and figure generation. Alpha-diversity (intra-sample) and beta-diversity (inter-sample) were calculated in Explicet at the rarefaction point of 72,603 sequences with 100 bootstrap re-samplings.
The first question targeted by this study was whether hypotonic lysis preferentially selects for (i.e., preferentially retains or lyses) particular groups of microbes. After the first hypotonic lysis, re-suspended pelleted cells were subsampled from the first two samples (CF1-1A* and CF1-2A*) to compare with the same samples after the second hypotonic lysis (CF1-1and CF1-2). All samples were treated equally, i.e., treated with DNase I prior to DNA extraction, followed by DNA extraction and the sequencing pipeline. As shown in Figure 4, the microbial profiles of the subsamples are highly similar to the samples after two hypotonic lysis treatments. In addition, the second hypotonic lysis increases the fraction of non-human sequences by 6-17% within the metagenomes (Table 2).
To test for differences in microbial composition between metagenomic- and 16S rDNA-based profiling, and for changes before and after hypotonic lysis that might explain the differences previously seen between our studies and others, bacterial 16S rDNA sequencing libraries were generated from both the total DNA and HL-derived DNA (Figure 4B). At genus level, the taxonomy profiles of the common CF-associated bacteria such as Pseudomonas, Stenotrophomonas, Prevotella, Veillonella,and Streptococcus were highly similar between the 16S rDNA libraries and metagenomes generated from the HL-derived DNA. However, Rothia detection in the 16S rDNA libraries was not as abundant as with the metagenomic libraries. When comparing the 16S rDNA taxonomical profiles generated from total DNA and HL-derived DNA, Pseudomonas was differentially represented in the total DNA compared to the HL-derived DNA starting from Day 3.
Metatranscriptomes
Typically, the total RNA extracted from CF sputum is partially degraded and the size ranges from 25-4,000 bps (Figures 5A and 5C). Here, the representative results presented was previously published in Lim et al. 20129. The fraction of rRNA within the non-depleted metatranscriptomes ranges from 27-83%, and the relative abundance of rRNA varied across samples (Table 3; data extracted from Lim et al.9). However, depletion with Ribo-Zero kit decreased the rRNAs relative abundance of rRNA to 1-5% with the exception of sample CF1-F. The variation in the effectiveness of rRNA removal could reflect the quality of extracted RNA, or differences in the microbial community present and hence the accessibility of rRNAs for probes hybridization9. The electropherograms of a successful (Figure 5B) and unsuccessful (Figure 5D) rRNA removal procedure using the Ribo-Zero rRNA removal kit differ, at which rRNA peaks are visible in the unsuccessful removal.
The size range of cDNA libraries generated often reflects the size range of the starting RNA sample. The cDNA libraries presented here were generated with a whole transcriptome amplification kit (WTA2) upon rRNA depletion followed by Roche-454 sequencing library preparation9. The cDNA generated contain fragments ranging from 50-4,000 bps (Figures 5E and 5F) and is highly consistent across samples (Lim et al. 2012)9. The availability of other platform-specific RNA-Seq library preparation kits currently provide more alternative options for one to combine cDNA synthesis and sequencing library preparation in optimum conditions. One recommended option to date is the ScriptSeq Complete Gold Kit combining rRNA removal reagents recommended above and RNA-Seq library preparation kit.
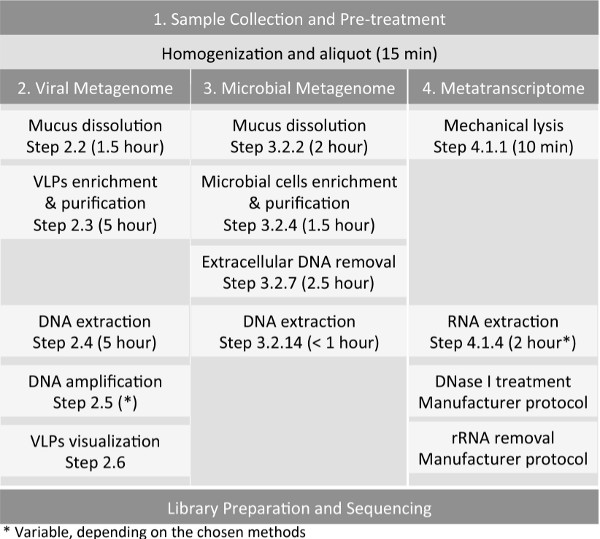
Figure 1: Workflow for the preparation of host-associated samples, such as sputum sample, for virome, microbiome, and metatranscriptome sequencing.
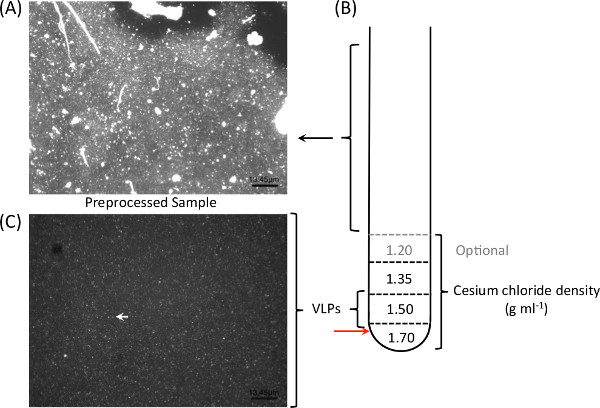
Figure 2: Cesium chloride density gradients ultracentrifugation facilitate the elimination of extracellular DNA and large particles (A), and allow for optimal isolation of viral-like particles from CF sputum. One milliliter of each gradient is layered on top of each other prior to loading the pre-treated sample (B). Following particles isolation and purification, epifluorescence microscopy with nucleic acid dyes such as SYBR Gold are used to verify the presence and purity of viral particles in samples. Clear viral-like particles (C; white arrow) were observed following the density gradient separation of CF sputum sample.
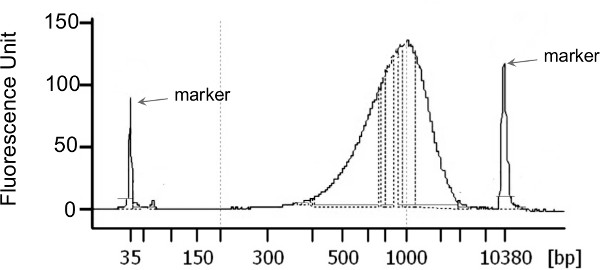
Figure 3: Example of the size distribution of Nextera XT libraries generated from 1 ng of HL-DNA that resulted in CF sputum microbiomes. Library normalization, pooling, and loading amount was done as described in the manufacturer protocol without any deviation.
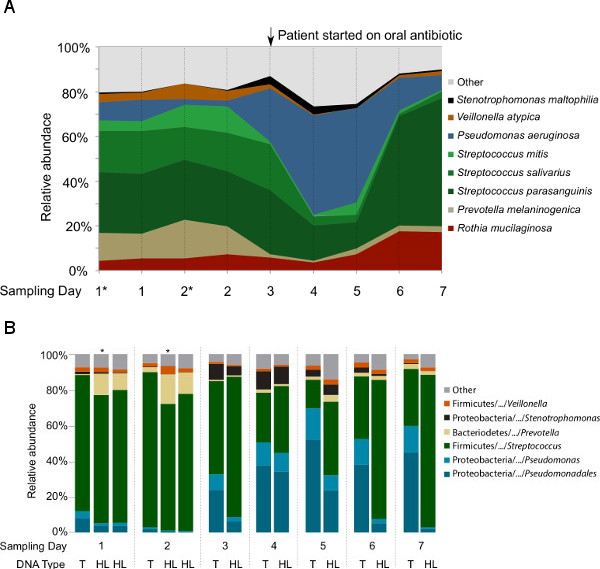
Figure 4: Taxonomic analysis of the microbial communities in nine samples collected longitudinally from one CF patient. (A) Microbial profiles based on the metagenomic libraries generated from hypotonic lysis method-based DNA. The species assignment was based on the Metaphlan pipeline following data preprocessing that remove duplicates and sequences with low quality and human sequence homology. In order to show that two-steps hypotonic lysis did not preferentially selects for particular groups of microbes, subsamples (*) after the first hypotonic lysis were included. (B) Microbial profiles based on the V1V2 region of 16S rRNA gene sequencing from total DNA (T) and hypotonic lysis method-based DNA (HL). These data have not been previously published.
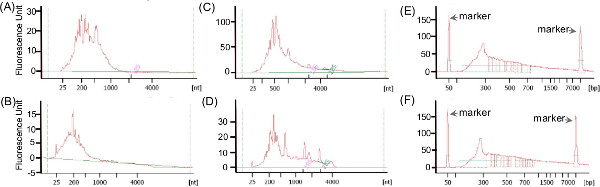
Figure 5: Examples of Agilent 2100 Bioanalyzer electropherograms of RNA (A-D) and cDNA (E-F) generated for the metatranscriptomic libraries, using RNA pico and high-sensitivity dsDNA chips respectively. (A) and (C) show the examples of electropherograms before rRNA removal procedures. The electropherograms of a successful (B) and unsuccessful (D) rRNA removal procedure using total rRNA Removal kit differ slightly, at which rRNA peaks are visible in the unsuccessful removal. The size range of cDNA (E-F) generated using the whole-transcriptome amplification kit (Sigma-Aldrich) is similar to the size range of the starting rRNA-depleted RNA, and highly consistent across the two different samples. Please click here to view a larger version of this figure.
| CF1-D | CF1-E | CF1-F | CF4-A | CF4-B | CF4-C | CF5-A | CF5-B | |
| Total number of reads | 224,859 | 87,891 | 106,189 | 93,301 | 140,020 | 1,558 | 272,552 | 217,438 |
| Preprocessed readsa | 109,389 | 73,624 | 67,070 | 82,011 | 68,617 | 1,137 | 215,808 | 158,432 |
| 49% | 84% | 63% | 88% | 49% | 73% | 79% | 73% | |
| Number of bases | 47,239,573 | 33,351,525 | 28,922,479 | 27,667,695 | 29,386,841 | 243,986 | 95,205,805 | 69,581,811 |
| Mean read length | 432 | 453 | 431 | 337 | 428 | 215 | 441 | 439 |
| Host sequencesb | 240 | 526 | 28 | 79,774 | 13 | 797 | 585 | 5,859 |
| 0.21% | 0.71% | 0.04% | 97.27% | 0.02% | 70.10% | 0.27% | 3.70% | |
| Viral hitsc | 7,214 | 23,550 | 4,070 | 737 | 4,642 | 22 | 6,466 | 5,981 |
| 6.59% | 31.99% | 6.07% | 0.90% | 6.77% | 1.93% | 3.00% | 3.78% | |
| Unassigned Readsd | 103,888 | 60,490 | 32,780 | 1,935 | 68,440 | 311 | 105,612 | 119,551 |
| 94.97% | 82.16% | 48.87% | 2.36% | 99.74% | 27.35% | 48.94% | 75.46% | |
| a Reads after data pre-processing by PRINSEQ29. | ||||||||
| b Human reads identified by DeconSeq30 plus reads with a best BLASTn hit (NCBI nucleotide database) to the phylum Chordata. | ||||||||
| c tBLASTx hits against in-house viral genome database. The percentage was calculated using the total number of preprocessed reads. | ||||||||
| d Reads with no BLASTn hit against the NCBI nucleotide database. The percentage was calculated using the total number of preprocessed reads. Some reads with no BLASTn hit against the NCBI nucleotide database were identified as viral at protein level in the tBLASTx analysis. | ||||||||
Table 1: Library characteristics of eight viromes generated from sputum samples using presented workflow. This table is extracted from Lim et al. (2012)9. Seven samples (CF1-D, CF1-E, CF1-F, CF4-B, CF4-C, CF5-A, and CF5-B) were processed as described in Section 2 and generated viromes that contained little (0.02% – 3.7%) human-derived sequences with one exception (70%). CF4-A was omitted from the density gradient ultracentrifugation step (CF4-A) and generated virome that contained > 97% human-derived sequences.
| Sample | Concentration | Total Yield | Total No. Reads | Total No. Reads (Processedb) | Non-human Sequences |
| (ng/μl) | (ng) | (Rawa) | (%) | ||
| CF1-1A* | 2.3 | 230 | 1,098,454 | 937,688 | 691,541 |
| 74% | |||||
| CF1-1 | 13 | 1,300 | 2,212,756 | 1,958,910 | 1,574,520 |
| 80% | |||||
| CF1-2A* | 2.1 | 210 | 672,878 | 588,106 | 407,530 |
| 69% | |||||
| CF1-2 | 5.2 | 520 | 1,944,012 | 1,697,010 | 1,455,174 |
| 86% | |||||
| CF1-3 | 28.8 | 2,880 | 1,048,304 | 896,756 | 560,852 |
| 63% | |||||
| CF1-4 | 24.1 | 2,410 | 1,154,922 | 984,702 | 621,098 |
| 63% | |||||
| CF1-5 | 33.6 | 3,360 | 1,029,622 | 888,630 | 481,548 |
| 54% | |||||
| CF1-6 | 43.2 | 4,320 | 1,434,016 | 1,256,504 | 725,858 |
| 58% | |||||
| CF1-7 | 57.8 | 5,780 | 1,000,174 | 872,036 | 565,376 |
| 65% | |||||
| * 1 ml of sample was subsampled from CF1-1 and CF1-2 following the first hypotonic lysis step (Step 3.1.5) before the second hypotonic lysis procedure. The cells were spun down as described in 3.1.7 and proceed through the remaining protocol without any modification. | |||||
| a Unprocessed Illumina reads from a 2 x 300 bp MiSeq sequencing run. | |||||
| b Reads were assessed, trimmed, and removed based on quality and length as described in the discussion. | |||||
Table 2: Characteristics of microbiomes generated from sputum samples using presented workflow. The DNA concentration of each sample in 100 μl elution buffer (5 mM Tris/HCl, pH 8.5) and the characteristics of sequence data are presented. A total of 1 ng was used to generate individual library using the Nextera XT library preparation kit.
| Sample | CF1-D | CF1-F | CF4-B | CF4-C | ||||
| Treatment | None | Ribo-Zero | None | Ribo-Zero | None | Ribo-Zero | None | Ribo-Zero |
| Preprocessed reads | 2,088 | 1,991 | 40,876 | 25,238 | 19,728 | 32,737 | 31,791 | 36,172 |
| Mean read length | 275 | 245 | 262 | 270 | 233 | 259 | 240 | 267 |
| Total rRNA reads | 1,737 | 91 | 29,499 | 17,267 | 5,285 | 291 | 16,371 | 1,761 |
| 83.20% | 4.60% | 72.20% | 68.40% | 26.80% | 0.90% | 51.50% | 4.90% | |
| Microbial rRNA | 1,414 | 32 | 19,978 | 12,035 | 23 | 227 | 6,916 | 1,076 |
| 67.70% | 1.60% | 48.90% | 47.70% | 0.10% | 0.70% | 21.80% | 3.00% | |
| Eukaryota rRNA | 323 | 59 | 9,520 | 5,232 | 5,262 | 64 | 9,455 | 683 |
| 15.50% | 3.00% | 23.30% | 20.70% | 26.70% | 0.20% | 29.70% | 1.90% | |
| % rRNA removed* | 0% | 95% | 0% | 5% | 0% | 97% | 0% | 91% |
| Non-rRNA reads | 351 (16.8%) | 1,900 (95.4%) | 11,377 (27.8%) | 7,971 (31.6%) | 14,443 (73.2%) | 32,446 (99.1%) | 15,420 (48.5%) | 34,411 (95.1%) |
| Total NR hits | 102 (4.9%) | 691 (34.7%) | 3,327 (8.1%) | 2,857 (11.3%) | 4,938 (25.0%) | 10,751 (32.8%) | 5,905 (18.6%) | 15,766 (43.6%) |
| Eukaryotic | 74 | 407 | 2,790 | 2,524 | 4,614 | 10,227 | 4,553 | 8,274 |
| Bacterial | 26 | 283 | 520 | 312 | 287 | 471 | 1,326 | 7,442 |
| Unassigned reads | 249 (11.9%) | 1,209 (60.7%) | 8,050 (19.7%) | 5,114 (20.3%) | 9,505 (48.2%) | 21,695 (66.3%) | 9,515 (29.9%) | 18,645 (51.5%) |
| *The amount of rRNA removed expressed as a percentage of the amount present in the non-depleted aliquot. | ||||||||
Table 3: Library characteristics of the metatranscriptomes with and without rRNA depletion. The data is extracted from Lim et al. (2012)9, which has additional comparison of other rRNA removal kits and the effect of cDNA nebulization prior to sequencing library preparation.
