Three 3D time-lapse recording experiments from three different C. elegans strains illustrate the type of data that can be obtained using the light sheet microscope setup described above. We checked that under these conditions the embryos develop at normal speed and survive the imaging, consistent with earlier reports indicating that SPIM imaging causes only low phototoxicity levels in C. elegans embryos6,7.
The first strain expresses an histone fused to GFP (zuIs178;stIs10024). Recording was performed during 2 hours with a stack of 20 slices (distance between slices 1 μm) taken every 37 sec. Figure 6 illustrates one plane of the stack at different time points. Both interphasic nuclei (arrow) and condensed mitotic chromatin (arrow head) are clearly visible.
The second strain expresses a tubulin fused to GFP (ruIs57) and an histone fused to mCherry (itIs37; stIs10226). Recording was performed during 16 min with a stack of 20 slices (distance between slices 1 μm) taken every 105 sec. Figure 7 illustrates different planes of the stack and different time points. The associated movie 1 displays 3D reconstructions at 3 successive time points. During division, the mitotic spindle (green) and condensed chromosomes (red) are clearly visible allowing the tracking of the cell division orientations during development.
The third strain expresses the apolipoprotein VIT-2 fused to GFP (pwIs23) and a membrane bound mCherry (ltIs44). Recording was performed during 25 min with a stack of 10 slices (distance between slices 1 μm) taken every 30 sec. Figure 8 illustrates different planes of the stack and different time points. The fast movements of the yolk lipoprotein particles (green) within the cells can easily be followed.
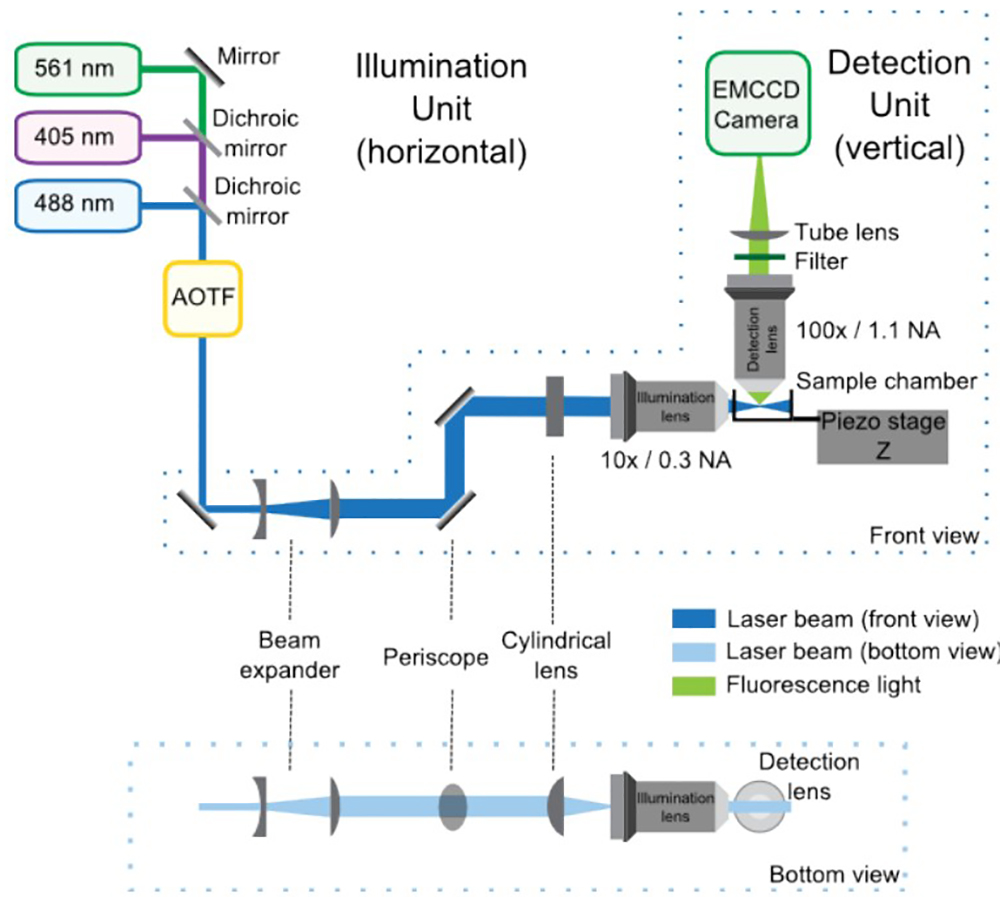
Figure 1: SPIM optical setup composed of the illumination and detection units, front and bottom views. In the illumination unit, the lasers combined by the dichroic mirrors enter the AOTF, which controls the power of each laser independently. Then, the telescope increases the size of the beam by 4-fold and the periscope brings it to the height of the microscope. The cylindrical lens forms the light sheet, which is refocused by the illumination objective. The detection unit is integrated in the upright microscope and is mainly composed by the detection lens, the filter, the tube lens and the EMCCD. The sample is positioned at the intersection between the illumination and detection paths. A piezoelectric stage allows vertical (Z) displacements of the sample for 3D acquisition.
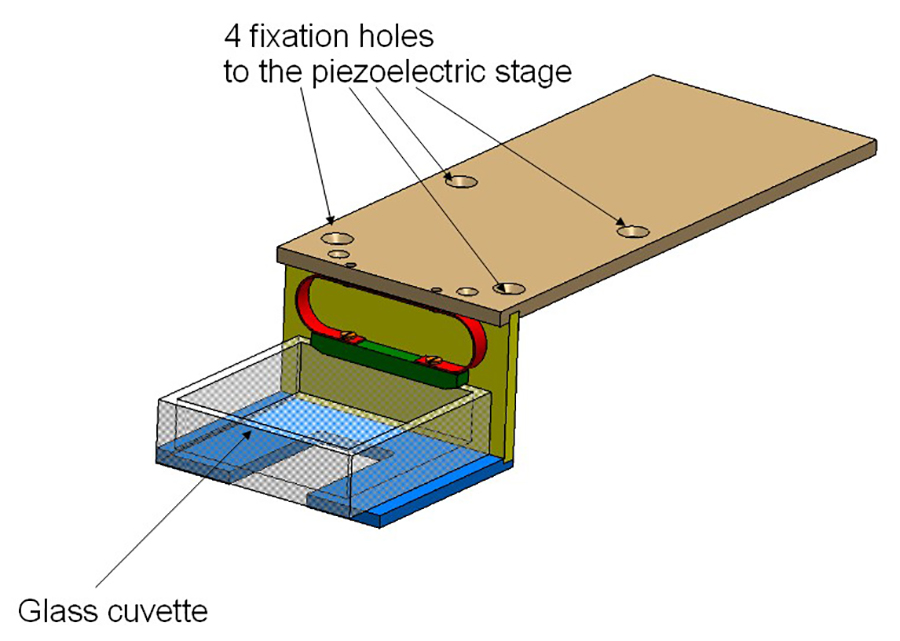
Figure 2: Cuvette holder. The holder consists of 3 aluminum plates and is screwed on the piezoelectric stage. The glass cuvette is held by a spring (in red).
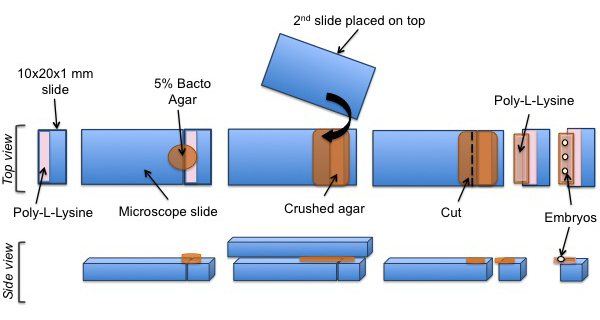
Figure 3: Mounting protocol of C. elegans embryos for SPIM experiments. A cut piece of glass is coated with poly-L-lysine. A pad of agar is positioned on the coated surface. Poly-L-lysine is then added onto the agar pad. Finally, C. elegans embryos are aligned on the poly-L-lysine coated agar pad.
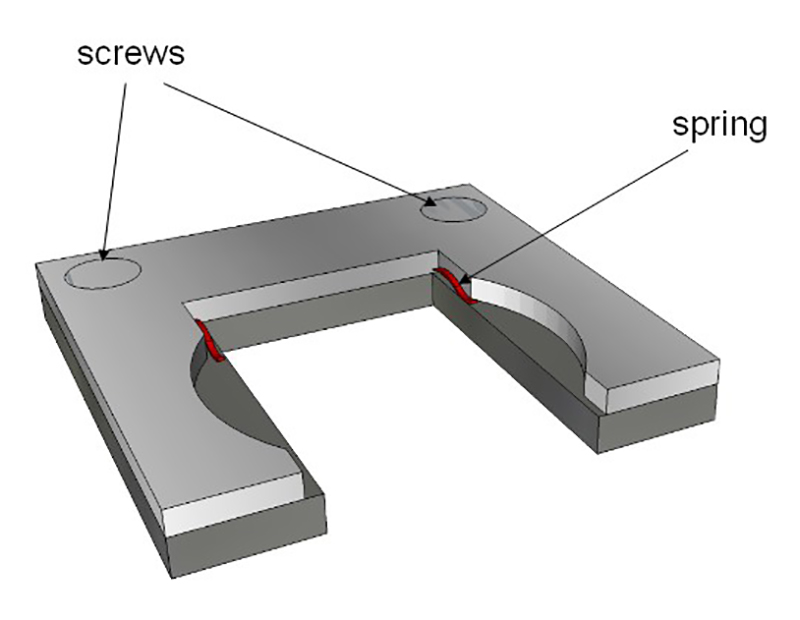
Figure 4: Sample holder. The holder fits to the interior dimensions of the glass cuvette. Two springs (in red) hold the glass slide (described in Figure 3). This holder is then put inside the glass cuvette.
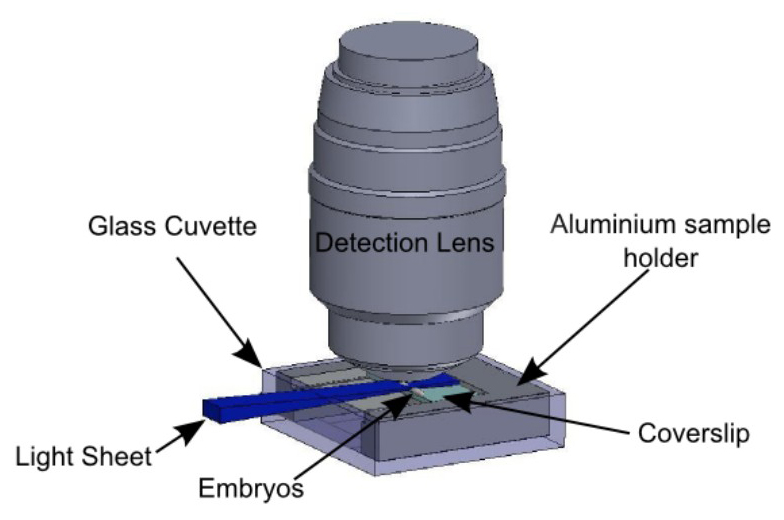
Figure 5: Sample holder in the observation context. Embryos are immobilized on the glass slide according to the protocol summed up in Figure 3. The slide is held by the sample holder described in Figure 4. The sample holder is inserted in the glass cuvette, held by the holder described in Figure 2. The light sheet is horizontal and illuminates the embryos from the side. The detection objective lens is vertical, above the sample.
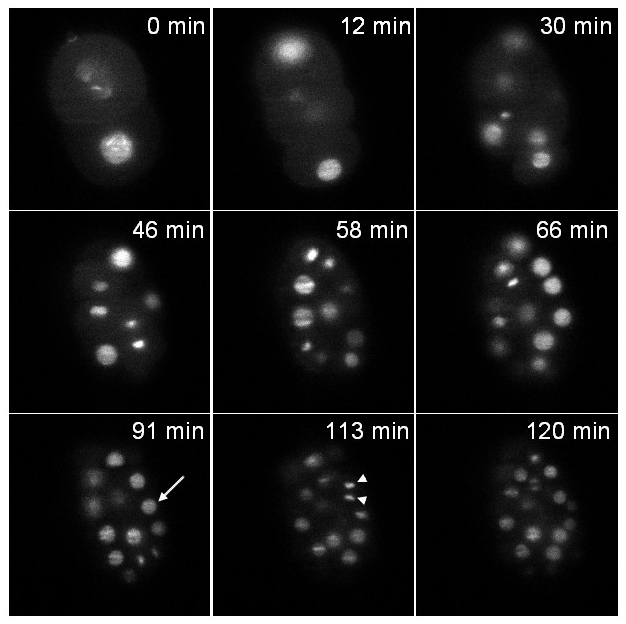
Figure 6: Recording of a C. elegans embryo expressing an histone::GFP fusion. Single plane images of a transgenic embryo expressing an histone::GFP fusion (zuIs178; stIs10024) at different time points. Images extracted from a 3D time-lapse recording: stacks made of 20 slices (distance between slices 1 µm); exposure time per slice: 30 msec; time interval between stacks: 37 sec; total acquisition time: 2 hr. Arrow: interphasic nucleus, arrow head: condensed mitotic chromatin. At t = 0 min the embryo is at the 2-cell stage and at t = 120 min the embryo contains around 70 cells as expected under normal developmental speed (note that only the cells contained in a single plane are visible in the pictures and not all the cells of the embryo). The power of illumination was 30 µW at 488 nm (measured at the exit of the illumination objective). Please click here to view this video.
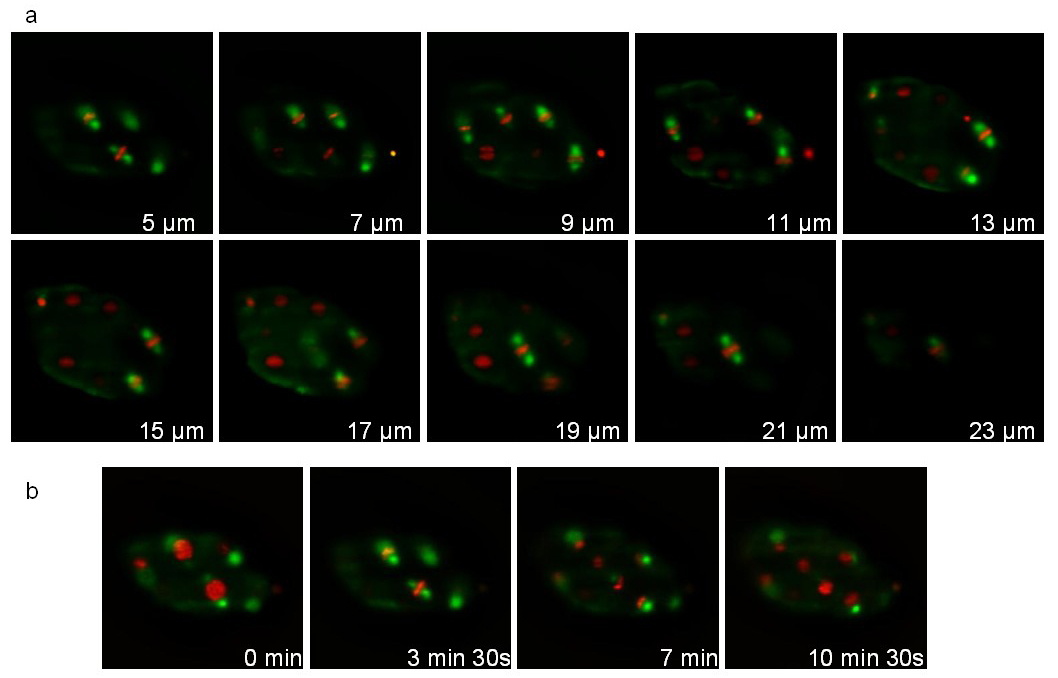
Figure 7: Recording of a C. elegans embryo expressing a Tubulin::GFP fusion and an Histone::mCherry fusion. Images extracted from a 3D time-lapse recording of a C. elegans embryo expressing a Tubulin::GFP fusion (ruIs57) and an Histone::mCherry fusion (itIs37 ; stIs10226). Recording conditions: acquisition of a stack of 20 slices (distance between slices 1 µm) every 105 sec; total acquisition time: 16 min; exposure time: 200 msec for each channel. Panel a shows 10 slices of the same stack. Panel b shows the same slice every 210 sec. The power of illumination was 30 µW and 300 µW, for 488 and 561 nm lasers, respectively (measured at the exit of the illumination objective). Please click here to view this video.
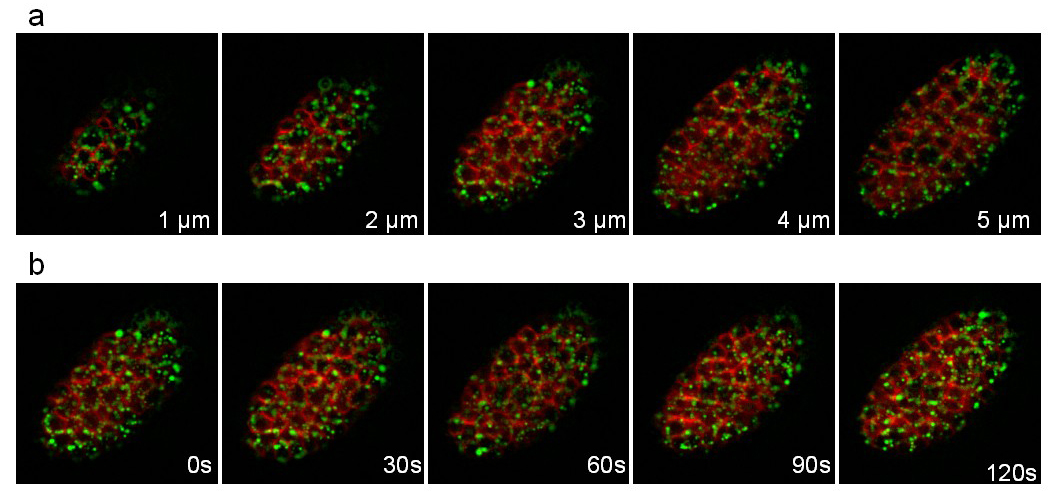
Figure 8: Recording of a C. elegans embryo expressing a VIT-2::GFP fusion and a membrane-targeted mCherry. Images extracted from a 3D time-lapse recording of a C. elegans embryo expressing a VIT-2::GFP fusion (pwIs23) and a membrane-targeted mCherry (ltIs44). Recording conditions: acquisition of a stack of 10 slices (distance between slices 1 µm) every 30 sec; total acquisition time: 25 min; exposure time per channel: 200 msec. Panel a shows the 5 first slices of the same stack. Panel b shows the same slice every 30sec. Please click here to view this video.
