1. Preparing Plates
- Use sterile technique at all times.
- Prepare fibronectin (FN) by diluting 100 μL of human plasma FN stock into a final volume of 3.3 mL in Dulbecco’s PBS (dPBS). Final concentration is 30 μg/mL and this can be stored at 4 °C for 1 week.
- Cover bottom of each well of a sterile tissue culture four well plate with FN solution and let sit for 15 minutes. Make sure the entire surface is covered. Make media during this time (steps 2 and 3).
- Remove FN solution and allow plates to dry. Gently rinse wells with 500 μL DMEM, remove DMEM, and add 500 μL of Self-Renewal (SR) medium (see below). Incubate at 37 °C in a humidified incubator containing 5 percent CO2. Complete this approximately one hour before dissection so that the plates will be dry before use.
2. Preparing SR Medium
- For the culture of vagal and trunk NC from ten embryos (approximately one litter, depending on genetic background of the mouse line), prepare 25 mL of SR medium. Combine 12.5 mL low glucose DMEM, 7.5 mL Neurobasal Medium, 25 μL retinoic acid (117 μM final concentration), and 25 μL 2-mercaptoethanol (50 mM final concentration). Mix well.
- Add 3.75 mL Chick Embryo Extract, 250 μL N2 salt supplement, 500 μL B27 supplement and 250 μL penicillin-streptomycin (1% final concentration). Filter the medium through a 0.22 μm filter.
- Add 10 μL sterile IGF1 (20 μg/mL final concentration) and 20 μL sterile bFGF (20 μg/mL final concentration). Mix by inverting. Store at 4 °C.
3. Preparing Wash Medium
- For approximately 10 embryos prepare 50 mL of wash medium. Combine 50 mg BSA with 35 mL low glucose DMEM, 15 mL Neurobasal Medium, and 500 μL penicillin-streptomycin (1% final concentration). Sterile filter with a 0.22 μm filter.
4. Preparing Collagenase/dispase
- Add 50 μL of 100 mg/mL collagenase/dispase to 5 mL dPBS. Mix well.
- Syringe filter with a 0.2 μm filter and add 1.5 mL into each of three wells of a twelve well plate. Pipette approximately 1 mL wash medium in the remaining wells. Store entire plate on ice until ready to digest dissected tissue.
- Cut tips of a p20 and p1000 pipette filter tip with a sterile razor blade. Cut just below the beveled edge of the tip. The cut off p1000 will be used to transfer the whole embryo while the p20 will be used to transfer pieces of isolated tissue.
5. Isolating Vagal and Trunk Neural Tube from 9.5 dpc Embryos
- For timed pregnancies, dams with a vaginal plug are considered 0.5 dpc at noon the morning the plug is observed. Sacrifice and remove the uterus at 9.5 dpc.
- Remove the decidua from uterus and gently remove the embryo from the decidua. Once experienced with this protocol, isolate 3-4 9.5 dpc embryos at a time in sterile dPBS. Use sterile technique and sterilized dissection instruments. Instruments can be sterilized by autoclaving, heat sterilization or by incubation in ethanol.
- For anterior vagal NC: using insulin needles, cut the neural tube at the mid otic placode. Cut again at the posterior edge of the 4th somite. Trim tissue ventral to the neural tube to remove the pharyngeal arches and heart.
- For trunk NC: using insulin needles, remove the portion of the neural tube between somites 16-22 (or the last somite if embryos are developmentally earlier than the 22 somite stage).
- Keep the yolk sac and any remaining embryonic tissue for genotyping.
6. Removal of Non-neural Ectoderm and Mesoderm
- Place neural tube containing segments into collagenase/dispase at room temperature for 10 minutes. Immediately wash in wash medium.
- Return tissue to sterile dPBS. Using sterile insulin needles, gently remove the non-neural ectoderm from the tissue and separate the somites away from the neural tube. The last remaining parts of the mesoderm from somite tissue can be removed by triturating gently using a cut-down p20 tip. Be careful to not damage the neural tube. Observe this closely during trituration.
- Place the isolated neural tube through a second and third 30 second wash of wash medium.
- Wash once in SR medium, and place the isolated neural tube into the center of a FN-coated well that was prepared previously (Step 1.3). Humidify the hypoxia chamber with a dish of sterile water. Place the dish into the hypoxia chamber and flush the chamber with mixed gas to 3% O2 (use a tank containing a mixture of 1% O2, 6% CO2, 93% N2).
- Always handle the chamber extremely carefully to ensure that explants are undisturbed and remain in the center of the wells.
- Incubate at 37 °C. Summary of steps 5-6 is shown in Figure 1.
7. Removal of Neural Tube
- After 24 hours of incubation, remove the neural tube by gently teasing the edge of the neural tube away from the migrating cells using a sterile insulin needle. Remove and discard the neural tube from the medium using a sterile p20 cut as in step 6.2 and replace the medium with fresh SR medium (Figure 2). This is most easily done using an inverted microscope.
8. Representative Results
Following 24 hours of incubation at 37 °C in hypoxic conditions, NC cells have migrated away from the neural tube in a nearly pure population (Figure 3a). Sometimes, less than ideal cultures will not yield robust outgrowths. For example, it is possible that after 24 hours the neural tube will have curled up upon itself and the NC will not migrate away from the neural tube (Figure 3b). Occasionally the neural tube will not attach to the fibronectin coated plates.
In our experience, sub-optimal NC migration or problems with attachment of the neural tube can be adversely affected by normoxia conditions or concentration of the fibronectin, respectively. Enzymatic activity of the collagenase/dispase varies slightly by batch and digestion time must be adjusted appropriately, however, do not digest the tissue longer than fifteen minutes. Overdigestion of the neural tube containing tissue in collagenase/dispase will also result in deficient outgrowths. If somite tissue is not easily removed from the neural tube after incubation in collagenase/dispase, the neural tube can be incubated for longer than ten minutes. Sometimes the neural tube will not attach to the substrate. If this is the case, double-check the fibronectin concentration and the hypoxia conditions.
While normoxic conditions can be used to culture wild-type NC, hypoxic conditions more closely mimic the in vivo environment29,30. In our experience, hypoxic conditions became critical when culturing mutant NC. For example, when Foxd3 mutant NC were cultured in normoxic conditions, trunk NC had a greatly reduced cell outgrowth compared to controls. This disparity in outgrowth size was removed when the explants were cultured in hypoxic conditions (Figure 4). Furthermore, when wild-type neural tube explants were cultured in normoxia, the number of caspase-positive cells was greater then that of similar explants cultured in hypoxia (data not shown). By maintaining all NC culture in hypoxia, comparisons can more easily be made between dynamics of control and mutant cultures.
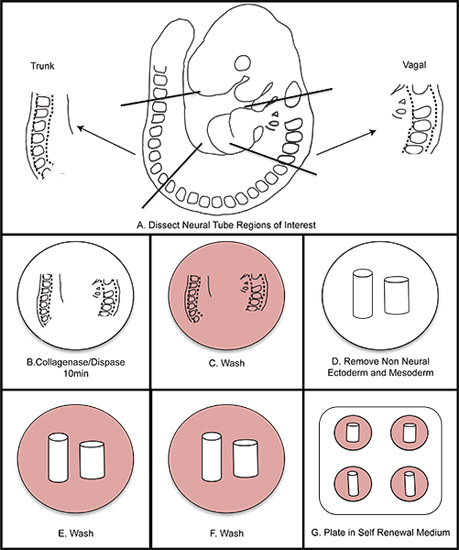
Figure 1. Overall schematic of NC isolation. A) Dissect regions of interest from the embryo. B) Digest neural tube in collagenase/dispase for ten minutes (do not exceed fifteen minutes). C) Wash in wash medium. D) Dissect away the non-neural ectoderm and mesoderm. E-F) Wash twice in wash medium. G) Plate in self renewal medium. Incubate at 37 °C in 3% O2 hypoxic conditions.
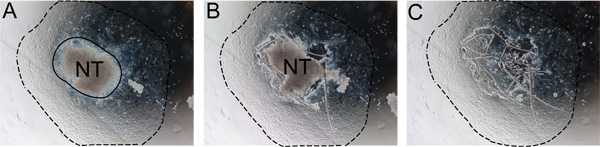
Figure 2. Stepwise removal of neural tube from explant. The neural tube must be removed after 24-48 hours to prevent contamination with non-NC cells. A) Notice the boundary between the neural tube and the NC outgrowth (solid line). B) Cut along the edge of the neural tube with an insulin needle. C) Discard the neural tube and replace medium with fresh self renewal medium. Dashed line indicates extent of the outgrowth. Abbreviation: NT, neural tube.
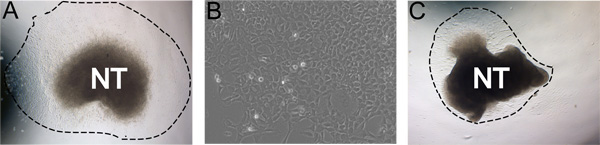
Figure 3. Examples of representative results. A) Typical explant outgrowth after 24 hours incubation in self renewal medium in hypoxic conditions (dashed line indicates extent of the outgrowth). B) Magnified view of NC outgrowth after 48 hours in culture. C) Less ideal culture with low outgrowth yield. A and C were cultured under the same conditions. Images demonstrate the natural range in culture robustness. This can be affected by efficiency of neural tube isolation, concentration of FN, hypoxic conditions, and time in collagenase/dispase digestions.
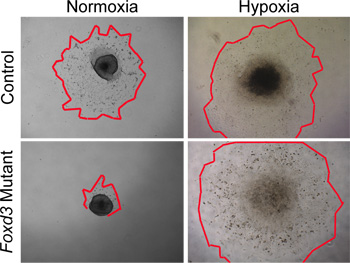
Figure 4. In vitro analyses of NC explant cultures in normoxia versus hypoxia. Control (wild type) NC cells migrated from neural tube explants after 48 hours in normoxic culture conditions. In contrast, Foxd3 mutant NC had greatly reduced cell outgrowths in normoxia (red outlines mark edges of the NC outgrowths). When comparable explants were grown under hypoxic conditions, Foxd3 mutant NC explants grew comparably to controls, allowing subsequent analyses. Note, this behavior correlated well with the behavior of Foxd3 mutant NC in vivo.
