Isolation of Stem Cells from Postnatal Mouse Cerebellum and Differentiation into Neural Cells
Abstract
Source: Edamakanti, C. R., et al. Purification of Prominin-1+ Stem Cells from Postnatal Mouse Cerebellum. J. Vis. Exp. (2020).
This video demonstrates a method for isolating stem cells from the postnatal mouse cerebellum and differentiating them into neural cells. Stem cells from the isolated cell suspension are purified using antibody-coated magnetic beads and are cultured to form neurospheres. The neurospheres are then maintained in a differentiation medium to generate neural cells.
Protocol
1. Preparation of Solutions
- Prepare tissue dissociation solution made from sterile phenol red-containing Dulbecco's phosphate-buffered saline (DPBS) with papain (100 U/ml), cysteine (0.2 mg/ml) and deoxyribonuclease (DNase, 250 U/ml).
- To prepare the DNase solution dilute 100 mg of the lyophilized powder of DNase I (one bottle) in 50 mL of H2O. Mix well and filter the stock solution. Prepare 10 mL stock aliquots. Divide one stock tube into 0.5 mL single-use aliquots. Store these single-use aliquots at -80 °C.
- Prepare magnetic separation reagents by preparing magnetic column buffer X: 0.5% bovine serum albumin (BSA) and 2 mM ethylenediaminetetraacetic acid (EDTA) solution.
- To prepare neurosphere media, use a neurobasal medium containing penicillin/streptomycin with L-glutamine and supplement with 2% B27, 20 ng/mL human recombinant epidermal growth factor (EGF), and 20 ng/mL human recombinant basic fibroblast growth factor (bFGF).
- To prepare differentiation media, use a neurobasal medium and supplement with 10 ng/mL differentiated factor platelet-derived growth factor (PDGF-AA) or 10 ng/mL leukemia inhibitory factor (LIF) and 2% B27.
- Use ultra-low attachment 12 well (3.5 cm2) and 6 well (9.6 cm2) culture plates.
2. Dissection of Cerebellum
- Anesthetize mouse pups (P3-P7) with isoflurane and decapitate using surgical scissors.
- Spray the separated heads of the pups with 70% ethanol.
- Transfer each head into an empty sterile 10 cm culture dish. Separate the skin using microdissection scissors, then remove the skull by running the scissors sagittal along the midline.
- Using #7 forceps, peel off the skull bones starting caudally from the brainstem. Carefully lift the brain using a spatula, keeping the cerebellum intact, and transfer the brain to a fresh sterile 10.0 cm dish containing 15 mL of ice-cold Hanks' Balanced Salt solution (HBSS) solution.
- Place the dish containing the brain under a dissection microscope. Using fine #5 forceps, remove the meninges and large blood vessels from the cerebellum and separate the cerebellum from the brainstem using the spatula.
- Transfer the cerebellum into a 15 mL centrifuge tube containing 5.0 mL of ice-cold HBSS solution. Wash the cerebellum by rinsing and decanting 3x with 5.0 mL of HBSS.
NOTE: Each cerebellum should be placed in its own tube for further processing.
3. Cell Suspension Preparation
- Place the dish containing the brain under a dissection microscope. Using fine #5 forceps, remove the meninges and large blood vessels from the cerebellum and separate the cerebellum from the brainstem using the spatula.
- Transfer the cerebellum into a 15 mL centrifuge tube containing 5.0 mL of ice-cold HBSS solution. Wash the cerebellum by rinsing and decanting 3x with 5.0 mL of HBSS.
NOTE: Each cerebellum should be placed in its own tube for further processing. - After the last rinse, add 5 mL of papain-based tissue dissociation solution that has been pre-warmed to 37 °C. Incubate the tissue for 15 min at 37 °C in a water bath. Slowly mix the contents by inverting the tube up and down 3x–5x every 3 min either using a nutating mixer or by hand.
- Prepare a wide diameter (regular glass pipette) and narrow-diameter Pasteur pipette (fire-polished by heating over a Bunsen burner) as described previously.
- Wash the tissue 3x with 5 mL of HBSS solution, avoiding loss of the tissue while decanting by hand.
- Remove the last HBSS wash and add 5 mL of DPBS solution containing 250 μL of DNase solution to the tissue. Dissociate the tissue by triturating 10x–15x using the wide-diameter Pasteur pipette. Perform this step gently to avoid the formation of bubbles.
- Then incubate the slurry for an additional 10 min at 37 °C in the water bath, mixing by inverting and straightening the tube.
- Use the reduced diameter Pasteur pipette to further triturate the tissue slurry 10x. If large pieces of tissue remain, press the tissue pieces against the bottom of the tube with the tip of the pipette gently and continue pipetting until the cells reach a fine suspension.
- Incubate the tissue at 37 °C for an additional 10 min, repeating the previous mixing steps.
4. Immunolabeling of Stem Cells
- Place the centrifuge tubes on ice and use ice-cold solutions for the next steps. Strain the dissociated cells through a 40 μm cell strainer into a 50 mL centrifuge tube. Top the filter with 10.0 mL of HBSS solution to ensure that cells pass through the mesh in this additional solution.
- Transfer the filtered cells into a fresh 15 mL centrifuge tube and centrifuge the cell suspension at 300 x g for 10 min at 4 °C. Aspirate and discard the supernatant completely by carefully using a vacuum aspirator.
- Resuspend the pellet in 160 μL of magnetic column buffer. To obtain the single-cell suspension before magnetic labeling, pass cells through a 30 μm nylon mesh to remove cell clumps, which can otherwise clog the column.
- Add 40 μL of anti-prominin-1 microbeads to each 15 mL tube, mix, and incubate in a refrigerator for 15 min in order for the antibody to bind to prominin-1-expressing cells.
- Wash the cells by adding 1.0–2.0 mL of column buffer X and centrifuge at 300 x g for 10 min. Aspirate the supernatant completely.
- Resuspend the pellet in 1.0 mL of column buffer X.
NOTE: If small clumps are seen after the resuspension of the pellet with 1.0 mL of column buffer X, they should be removed carefully using the tip of the Pasteur pipette, otherwise these clumps can block the magnetic column during cell sorting.
5. Magnetic Column Preparation, Cell Sorting, and Plating
NOTE: The magnetic separation from different genotype conditions (disease vs. control) must be performed at the same time, since any delay may affect the neurosphere morphology.
- Prepare the magnetic columns by placing them on the magnetic stand exposed to the magnetic field. Rinse the column once with 500 μL of buffer X by applying buffer that drips into a centrifuge tube to be discarded.
NOTE: Prepare fresh magnetic column buffer X for each experiment; if not, then stem cell yield will be low. - Apply the labeled cell suspension onto the column. Collect the flowthrough containing unlabeled cells that mainly consist of cerebellar neuronal/glial mixed cells into fresh 15 mL tubes (Figure 1A).
- Wash the column 3x with 500 μL of buffer X (each wash takes around 2–4 min).
NOTE: Cerebellar neuronal/glial mixed culture enriched with cerebellar granular neurons can serve as a useful byproduct of this purification step. - Remove the column from the magnetic field and place it in a 1.5 mL tube. Add 1.0 mL of culture medium (neurosphere medium) to the column and push the plunger into the column to flush out the cells tagged with prominin-1 beads into a fresh 1.5 mL falcon tube.
- To enhance the purity of prominin-1 labeled cells, pass the eluted cells over a second column following steps 5.1–5.4.
- Count the cells with a hemocytometer. The typical yield is 107 cells per cerebellum. Plate the cells onto ultra-low attachment plates (6 or 12-well, based on the density required for downstream experiments).
6. Passaging of Neurospheres and Differentiation
- Plate the prominin-1-labeled stem cells on ultra-low attachment 12 well plates in neurosphere medium (5,000 cells/well).
- After 7–10 days, the cells divide to yield ball-shaped floating neurospheres (primary neurospheres).
NOTE: In this experiment, secondary neurospheres that expand in numbers are generated for further use. Primary neurosphere populations are not used for these experiments since they may contain contaminating cells that do not have stem cell properties and can clump together with neurospheres. - For passaging, transfer the primary neurospheres along with the culture media using 1.0 mL pipette tips to a 15 mL sterile centrifuge tube. Pellet the neurospheres by centrifugation at 300 x g for 5 min and discard the supernatant.
- Resuspend the pellet in 5 mL of tissue dissociation media that contains papain or 0.05% trypsin solution. Incubate at 37 °C for 10 min.
- Centrifuge the cell suspension at 300 x g for 5 min. Resuspend the cells in 5 mL of neurosphere medium and dissociate the cells mechanically using a plastic Pasteur pipette and slowly pipetting up and down 10x.
- Plate the cells (again) in neurosphere media as described earlier. After 7–10 days in culture, the plate should be enriched with secondary neurospheres.
NOTE: These cultures can be passaged up to 8x with good efficiency, after which neurosphere size tends to be smaller and suggestive of a decrease in proliferation. - Count the cells on the hemocytometer and plate the optimal number for future experiments on poly-D-lysine coated plates (6 or 12 wells).
- To differentiate the stem cells, collect neurospheres from the second to eighth passage by centrifugation as described earlier, except add a differentiation medium to the pellet.
NOTE: After 7 days in vitro, the stem cells differentiate into neurons, astrocytes, and oligodendrocytes, as demonstrated by staining with the neuronal marker β-III tubulin, astrocytic marker Glial fibrillary acidic protein (GFAP), and oligodendrocyte marker O4.
Representative Results
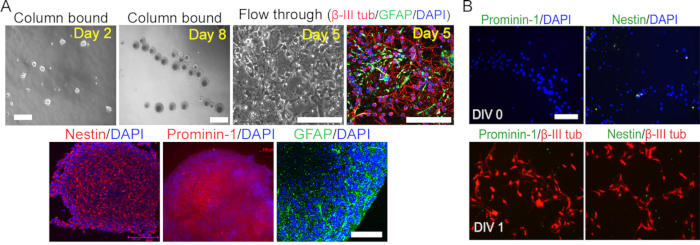
Figure 1: Isolation of prominin-1 stem cells from postnatal cerebellum. (A) Cerebellar stem cells were isolated using immunomagnetic prominin-1 beads. Top panel: Purified stem cells (column-bound) formed neurospheres with extensive proliferation and self-renewal properties. The cells unbound to the column (flowthrough) were unable to form neurospheres; instead, they became cerebellar neuronal/glial mixed cells (β-III tubulin/GFAP). Bottom panel: the neurospheres formed from prominin-1+ cells expressing stem cell-specific markers: Nestin, prominin-1, and GFAP. (B) Cells in the flowthrough stained negative for stem cell markers prominin-1 and nestin and positive for neuronal marker β-III tubulin.
Divulgazioni
The authors have nothing to disclose.
Materials
| 0.05% Trypsin | Thermo Fisher Scientific | 25300054 | 0.05% |
| 2% B27 | Gibco; Thermo Fisher Scientific | 17504001 | |
| 2mM EDTA solution | Corning | 46-034-CI | |
| Anti- Prominin-1 microbeads | Miltenyi Biote | 130-092-333 | |
| Bovine serum albumin | Sigma | A9418 | |
| Column MultiStand | Miltenyi Biotec | 130-042-303 | |
| culture plates ultra – low attachment | Corning | 3473 | |
| cysteine | Sigma | C7880 | |
| DNase | Sigma | D4513-1VL | 250 U/ml |
| Dulbecco's Phosphate Buffer Saline | Thermo Fisher Scientific | 14040141 | |
| Hank's balanced salt solution-HBSS | Gibco | 14025-092 | |
| Human recombinant Basic Fibroblast Growth Factor | Promega | G507A | 20 ng/ml |
| Human recombinant Epidermal Growth Factor | Promega | G502A | 20 ng/ml |
| Leukemia Inhibitory Factor | Sigma | L5158 | |
| l-glutamine | Gibco | 25030081 | |
| Microscopy | Lieca TCS SP5 confocal microscopes | ||
| MiniMACS separator | Miltenyi Biotec | 130-042-102 | |
| mouse anti-Prominin-1 | Affymetrix eBioscience | 14-1331 | 1 in 100 |
| Nestin | Abcam | ab27952 | 1 in 200 |
| Neurobasal medium | Thermo Fisher | 25030081 | |
| O4 | Millopore | MAB345 | |
| Papain | Worthington | LS003126 | (100 U/ml) |
| Platelet- Derived Growth Factor | Sigma | H8291 | 10 ng/ml |
| Poly-D-Lysine | Sigma | P6407 | |
| rabbit anti-tubulin, b-III | Sigma | T2200 | 1 in 500 |
| Rabit anti-GFAP | Dako | Z0334 | 1 in 500 |
| Separation columns-MS columns | Miltenyi Biotec | 130-042-201 | |
| Sterile cell strainer | Fisher Scientific | 22363547 | 40um |
