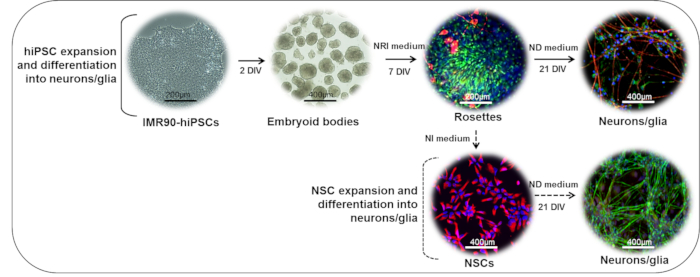
Figure 1: Schematic Representation of the Neuronal Differentiation Protocol. (Upper part) IMR90-hiPSC colonies can be cut into fragments to form embryoid bodies (EBs). After 2 days in vitro (DIV), EBs can be plated onto laminin- or standard matrix-coated dishes and cultured in the presence of neuroepithelial induction (NRI) medium to generate neuroectodermal derivatives (rosettes, here stained for nestin (green) and β-III-tubulin (red)). Rosettes can be dissociated, collected, replated on laminin- or standard matrix-coated dishes, and further differentiated into mature neuronal (NF200, red) and glial (GFAP, green) cells in the presence of neuronal differentiation (ND) medium. (Lower part) rosette-derived NSCs (nestin, red) can be expanded in the presence of neural induction (NI) medium, cryopreserved, or further differentiated in the presence of ND medium to form mixed neuronal (NF200, green) and glial (GFAP, red) cultures.
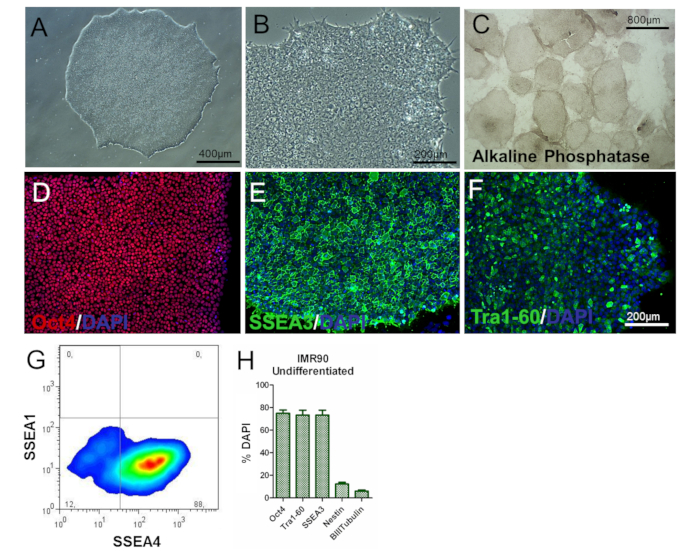
Figure 2. Characterization of Undifferentiated IMR90-hiPSCs. (A and B) Representative phase-contrast images (10X and 20X magnifications) of undifferentiated IMR90-hiPSC colonies. (C) representative images of alkaline phosphatase-stained colonies (4X magnification). (D-F) Representative immunocytochemical images of (D) Oct4 (red), (E) SSEA3 (green), and (F) Tra1-60 (green). (G) Representative dot plot of SSEA1 (CD15) and SSEA4 staining, analyzed by flow cytometry. (H) The bar graph shows the percentages of Oct4+ (~75 – 80%), Tra1-60+ (~75 – 80%), SSEA3+ (~75 – 80%), nestin+ (~10 – 15%), and β-III-tubulin+ (~3 – 7%) cells, counterstained with DAPI and quantified by HCI, with a mean of 3 to 5 biological replicates ± the S.E.M.
