This culturing method is considered successful if 1) the proliferation of the cells is approximately consistent across the control groups (and ideally all experimental groups), 2) the proliferation is appropriate given the seeding density, length of treatment, and the doubling time of the cell type/line, and 3) the viability of the harvested cells is 85% or higher (Table 1). Ideally, the resulting cells should be as healthy as they would be in standard cell culture, especially for use in subsequent experiments and assays (i.e., viability 85% or higher). This culturing method is considered unsuccessful if the opposite is true, whereby the resulting cells either die, differ substantially in proliferation across the control groups, or have suboptimal viability around 70% or lower (Table 1). Proliferation in the SMG treatment group may or may not differ compared to the controls, depending on how the SMG treatment affects the cellular physiology. However, this has not been an issue to date, and the cell proliferation was approximately equal across the control and treatment groups (Table 1). As mentioned previously, these parameters are important for the success of downstream assays and experiments, and the resulting cells from the two control groups should perform relatively similarly.
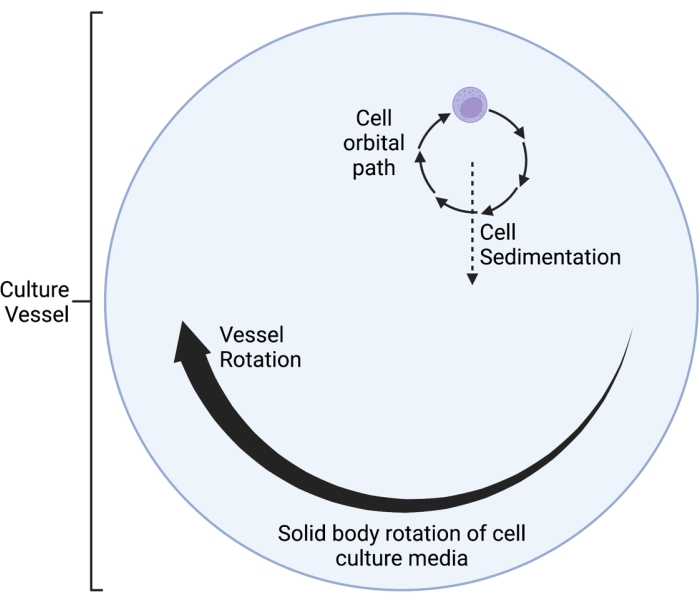
Figure 1: Schematic diagram of the localized orbital path of the cells cultured within the simulated microgravity (SMG) vessel during operation. The RWV 2D clinostat described here operates on the principle of time-averaged gravity vector nullification5,6,8,9, whereby the gravity vector is randomized through rotation of the cell culture on a horizontal axis. This is achieved by matching the rotational velocity of the culture vessel to the sedimentation velocity of the cells. After an initial speed-up phase, the media in the culture vessel eventually reaches "solid body rotation" over time. This horizontal rotation also induces laminar flow in the cell culture vessel. This creates a "low shear" environment, given that the shear stress induced on the cells by laminar flow is much less than that of turbulent flow. However, given that the clinostat is not a perfect system, there are some small, laminar fluid motions introduced, which inflict minimal shear stress on the cells. As such, the cells suspended in the media get dragged along by this flow during rotation. During horizontal rotation, the gravity vector acts on the cells and brings them into an oscillating trajectory, which is visualized here. As the culture vessel rotates on a horizontal axis, the gravity vector experienced by the cells rotates as well. Over time, this rotating gravity vector averages to approach zero; this phenomenon is called "time-averaged gravity vector nullification," and induces a state of SMG5,6,8,9. This Figure has been modified from Castro et al., 201120. Created with BioRender.com. Please click here to view a larger version of this figure.
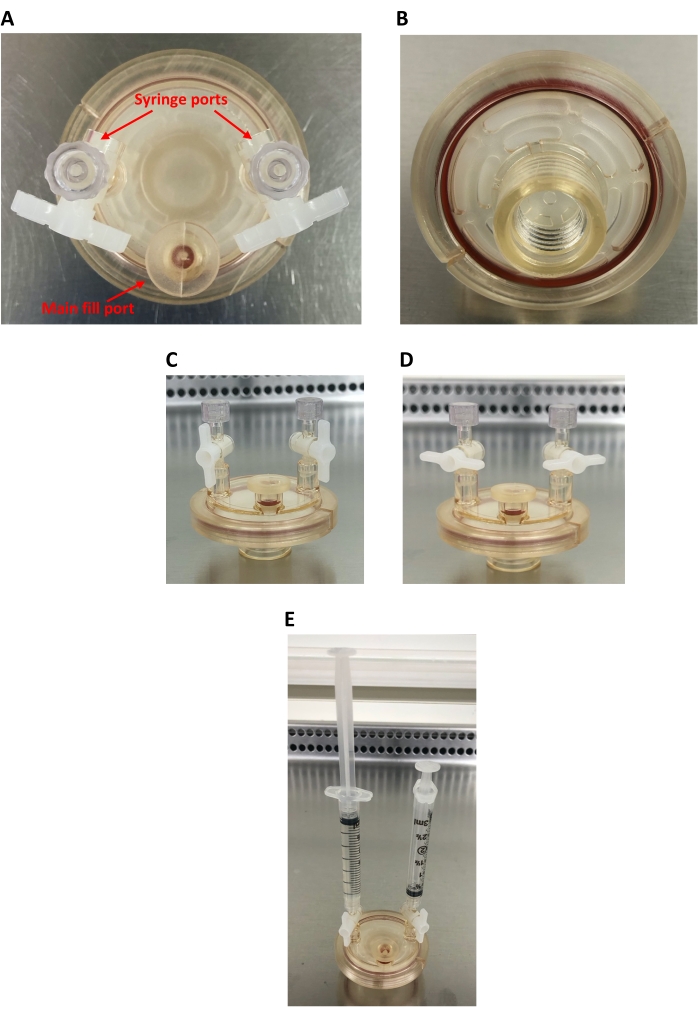
Figure 2: 10 mL high aspect ratio vessel (HARV). (A) Bird's-eye view of the HARV, showing the main fill port and two syringe ports. (B) Back of the HARV showing the screw-on port for connecting the HARV to the rotary base and the oxygenation membrane. (C) Side view of the HARV showing open syringe ports (capped). (D) Side view of the HARV showing closed syringe ports (capped). (E) Side view of the HARV showing the two 3 mL syringes attached; the left syringe is filled with cell culture and the right syringe is empty. Please click here to view a larger version of this figure.
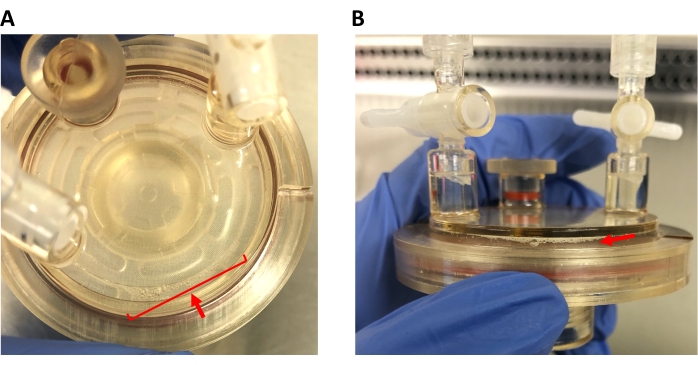
Figure 3: Bubbles in the HARV. (A) Bird's-eye view of the HARV, showing bubbles to be eliminated from the cell culture. (B) Side view of the HARV, showing the same bubbles. Note how the bubbles vary in size; microbubbles must be removed as well. Please click here to view a larger version of this figure.

Figure 4: Rotating base and power supply of the RWV device. (A) Front view of the rotating base, showing four rotating pegs that can accommodate up to four culture vessels. (B) Back view of the rotating base, showing the input for the ribbon cable (not pictured) that links the base and the power supply. (C) Front view of the power supply kept on top of the incubator. Note the on/off switch on the left and the rpm adjustment dial on the right. The power supply is plugged into the nearest outlet (usually on the back of the incubator) and includes an input for the ribbon cable to connect to the rotating base. The power supply stays outside of the incubator. The base is placed inside the incubator (37 °C, 5% CO2) during operation, and the ribbon cable is fed through the incubator door and connected to the power supply. The ribbon cable does not interfere with the incubator seal. When the rotating base is not in use, it should be kept outside of the incubator, safely stored on a lab bench or shelf. Refer to the Table of Materials for the details of the commercial device used. Please click here to view a larger version of this figure.
| # | Starting Viability | Seeding Density cells/mL | End Viability (F) | End Viability (1G) | End Viability (SMG) | End Conc (F) cells/mL | End Conc (1G) cells/mL | End Conc (SMG) cells/mL | Notes | |||||
| Suboptimal starting viability and seeding density (negative outcome) | 1 | 79% | 0.2 x 106 | 67% | 60% | 60% | 0.10 x 106 | 0.075 x 106 | 0.071 x 106 | Low seeding density and suboptimal starting viability led to cell death over the course of the treatment period | ||||
| 2 | 73% | 0.2 x 106 | 43% | 63% | 70% | 0.071 x 106 | 0.081 x 106 | 0.085 x 106 | ||||||
| Optimal starting viability and seeding density (positive outcome) | 3 | 93% | 0.4 x 106 | 93% | 93% | 96% | 1.2 x 106 | 1.1 x 106 | 1.5 x 106 | Appropriate seeding density and optimal starting viability led to healthy cell growth and viability throughout treatment | ||||
| 4 | 92% | 0.4 x 106 | 92% | 92% | 94% | 0.81 x 106 | 0.80 x 106 | 0.70 x 106 | ||||||
Table 1: Comparison chart showing unsuccessful and successful simulated microgravity (SMG) treatments. NK92 starting viability and seeding densities were compared to the resulting end viability and end concentrations after a 72 h SMG treatment in a 37 °C cell culture incubator supplemented with 5% CO2. Two instances of negative outcomes and two instances of positive outcomes were compared. For comparison, note that the optimal concentration range of the NK92 cell line used was between 0.3 x 106 cells/mL and 1.2 x 106 cells/mL, with a doubling time of around 2-3 days.
