Described below are the results of a successful Hi-C protocol (see a summary of the Hi-C protocol workflow in Figure 1A). There are several quality control checkpoints during the in-nucleus Hi-C experiment. Sample aliquots were collected before (UD) and after (D) the chromatin restriction step as well as after ligation (L). Crosslinks were reversed, and DNA was purified and run on an agarose gel. A smear of 200-1000 bp was observed when restriction with Mbo I was successful (Figure 1B). The expected size of the molecule depends on the restriction enzyme of choice. If the ligation was successful, a high molecular weight band was seen at the top of the gel (Figure 1B). Digestion efficiency can be also confirmed by qPCR as described in detail in the protocol. An acceptable digestion efficiency is 80% or higher (Figure 1C).
To assess Hi-C ligation efficiency in detail, primers can be designed to amplify an internal ligation product control in which the primers are in forward-forward or reverse-reverse orientation in adjacent restriction fragments. Alternatively, primers can be designed to amplify known interactions. Figure 2A shows the amplification of a known medium-range (300 kb) interaction in Drosophila25. Hi-C ligation products (in which the biotin marking, fill-in, and ligation occurred successfully) can be estimated by digestion of the PCR product recovered in the amplification. After fill-in and ligation, Hi-C amplicons will contain a new Cla I restriction site at the original Mbo I site, which is preserved upon blunt-end ligation. If restriction with Cla I is not complete, the fill-in reaction and biotin marking will be inefficient. A digestion efficiency of more than 70% is recommended to avoid having a large proportion of non-useful reads for the libraries after sequencing (Figure 2A, compare the Cla I digestion of the 3C versus the Hi-C template).
To determine an adequate number of PCR amplification cycles to amplify the final Hi-C library, PCR reactions were set up using 2.5 µL of a given library on beads, as described in the protocol. The number of PCR cycles for the final amplification is one cycle less than the number of cycles for which the smear is visible (Figure 2B). In this case, 4 cycles of PCR amplification were chosen. As a final quality checkpoint, an aliquot of the Hi-C library was re-amplified and digested with Cla I. The level of digestion of the library (a decrease in the smear size) indicates the abundance of valid Hi-C pairs and reflects the proportion of useful reads that will be obtained from the library (Figure 2C). A ratio of the upper size range (determined by the size present in the UD sample) and the bottom size range in both UD and D samples should produce a ratio > 1 for the UD and a ratio ≤ 1 for the digested sample if the Cla I digestion is efficient.
After paired-end sequencing, the FASTQ files (Table 1) were processed using HiCPro28 and the generated statistics plotted using MultiQC28. An alternative tool to HiCPro is the HiCUP30 pipeline that yields similar results (not shown). Figure 3 and Table 2 show the detailed statistical information of the sequenced reads. Full read alignment and alignment after trimming are reported. These two categories correspond to successfully aligned reads that will be used in subsequent analysis to find valid Hi-C pairs. The alignment-after-trimming category refers to reads spanning the ligation junction, which were not aligned in the first step and are trimmed at the ligation site to then realign their 5' extremity to the genome28 (Figure 3B,C and Table 2). The contact statistics show that the Hi-C library was of high quality with 82.2% valid pairs and 7.6% non-useful reads falling into the same-fragment self-circle, same-fragment dangling-ends, re-ligation, filtered pairs, and dumped pairs categories (Figure 3A, Figure 3D, and Table 2). Moreover, the number of PCR duplicates is very low, indicating that the library complexity is high, and that the PCR cycles introduced minimal artifacts (Figure 3E and Table 2).
Using the unique valid Hi-C pairs, basic analysis of the pair distribution was performed using HiCPro27. This experiment yielded 46.5% unique cis contacts ≤ 20 kbp, 47.1% unique cis contacts > 20 kbp, and 5.8% unique trans contacts (Figure 3D). The distribution of cis to trans valid pairs corresponded to the results expected for a successful Hi-C experiment with most of the interactions detected within the same chromosome. A high proportion of trans contacts indicates inefficient fixation. Using the Hi-C valid pairs from HiCPro27, matrices were normalized by iterative correction and eigenvector decomposition (ICE)30, and 1 kb and 5 kb resolution matrices were generated using HiCPlotter30,31,32. Normalized contact matrices at 1 kb and 5 kb resolution are presented for the Notch gene locus in Drosophila (Figure 4A, Figure 4C, and Figure 4D). In Figure 4A, the Notch gene locus can be seen along with the APs, domain l and II, as well as histone modifications along the locus (Figure 4A and Table 1). The design of the CRISPR-Cas9 deletion involved the motif of CTCF and M1BP (Figure 4B).
Upon deletion of the region containing both CTCF and M1BP DNA-binding sites at the 5' boundary of the Notch locus (5pN-delta343, Table 1), a dramatic change in chromatin contacts can be observed with loss of interactions inside the Notch locus and gain of contacts with the upstream TAD compared to the wild type (WT) (Figure 4C,D). Finally, Figure 4E shows a detailed panorama of WT and mutant interaction profiles at the restriction fragment level from the Notch gene 5' UTR, showing a decrease in the proportion of contacts made with the Notch gene locus and an increase in contacts with the upstream domain. The virtual 4C views of the Notch gene 5' UTR were obtained using HiC-Pro27. An alternative tool is the Hi-C other-ends quantification available in SeqMonk (SeqMonk (RRID:SCR_001913) http:www.bioinformatics.babraham.ac.uk/projects/seqmonk/. All the results presented in Figure 4 were obtained by applying the in-nucleus Hi-C protocol in WT and mutant S2R+ Drosophila cells, as described by Arzate-Mejía et al.25.
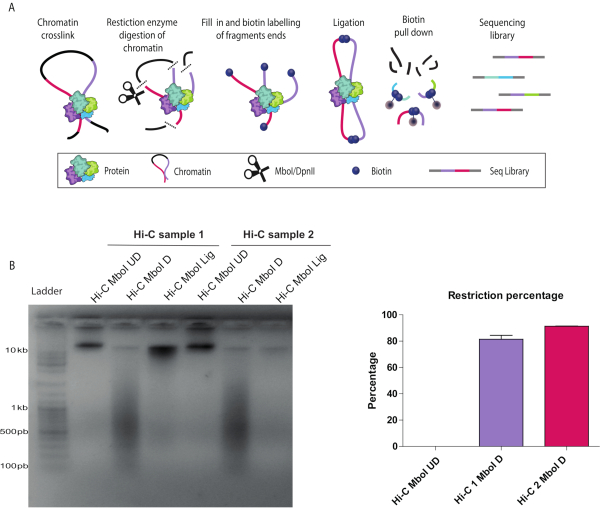
Figure 1: In-nucleus Hi-C digestion and ligation controls. (A) Hi-C protocol overview. Cells are cross-linked with formaldehyde, resulting in covalent links between chromatin segments (DNA fragments: pink, purple) and proteins. Chromatin is digested with a restriction enzyme (represented by scissors), in this example, Mbo I. The resulting sticky ends are filled in with nucleotides including a biotinylated dATP (Dark blue circles). DNA is purified, and the biotinylated junctions are enriched using streptavidin-coated magnetic beads (grey circles). Interacting fragments are identified by next-generation, paired-end sequencing. (B) Hi-C digestion and ligation quality controls for two biological replicates (Hi-C 1 and Hi-C 2). Hi-C libraries were resolved on a 1.5% agarose gel. Both digested, D, libraries run as a smear around 600 bp. Ligated samples, Lig, run as a rather tight band larger than 10 kb similar to the undigested UD samples. The differences in signal strength are due to uneven amounts of loaded DNA on the gel. (C) Hi-C digestion quantitative control by quantitative polymerase chain reaction for the same two biological replicates as in (B) (Hi-C 1 and Hi-C 2) using the cycle threshold values as detailed in the protocol. A successful digestion has ≥ 80% restriction. Please click here to view a larger version of this figure.
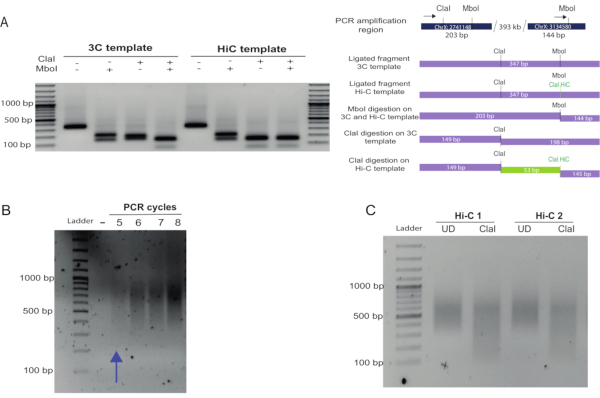
Figure 2: In-nucleus Hi-C fill-in and blunt-end ligation controls. (A) Fill-in and biotin labeling assessment. A known interaction between fragments located 300 kb apart in chromosome X was used as a control and amplified using the primers indicated with black arrows (see top of the scheme in (B), primer 1 (left), primer 2 (right), Table 1), generating a 347 bp amplicon. Hi-C ligation products can be distinguished from those produced in a 3C experiment by digestion of the ligation site. Hi-C junctions were digested by Cla I at the original Mbo I site, as this formed upon blunt-end ligation. Hi-C and 3C junctions were digested with Mbo I as the restriction site regenerates upon ligation (left of the gel). In contrast, 3C junctions were not digested by Cla I at the Mbo I site, but only by Mbo I. Compare the digestion profile of the Hi-C and 3C products using Cla I. A 53 bp fragment was obtained by digesting the Hi-C product (due to restriction of the Cla I site formed at the Mbo I site and restriction of a Cla I site already present in the region). This fragment was not observed in the 3C product digestion as the only Cla I site available was the one that was already present in the region. (B) After PCR amplification of the Hi-C library using different PCR cycles, the products were run on a 1.5% agarose gel. A smear of 400-1000 bp was expected and observed. The appropriate number cycles for the final amplification PCR should be taken as the number immediately lower than that at which a smear is just visible. (C) Final library Cla I digestion. An aliquot of the final library was re-amplified and digested with Cla I. The size reduction of the smear confirmed that a large proportion of the molecules in the library were valid Hi-C pairs. Densitometric analysis of this gel can be performed to obtain a ratio between the UN and D samples, as detailed in the representative results section. Please click here to view a larger version of this figure.
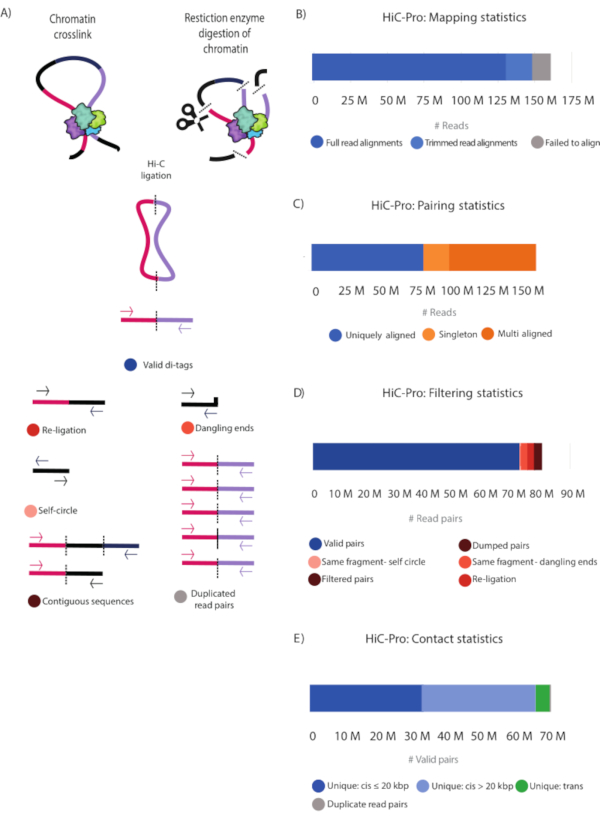
Figure 3: HiC-Pro statistics of the Hi-C library. (A) Schematic representation of valid Hi-C pairs and the different types of non-valid pairs that can be produced during the experiment and filtered out by HiCPro27 (Table 2). These include reads falling into contiguous sequences, dangling ends, same-fragment, self-circle, re-ligations, and PCR duplicates. (B) Mapping statistics. Reads that failed to align are shown (grey), and both fully aligned reads and reads aligned after trimming are shown in blue and light blue, respectively. These two categories represent the useful reads that are considered in subsequent analyses. (C) Pairing statistics. Multi Aligned reads (dark orange) represent reads that are aligned in multiple regions in the genome. Uniquely Aligned (dark blue) reads represent the read pairs that are aligned once in the genome, and singletons (light orange) represent read pairs in which just one genomic region was sequenced in both reads. (D) Filtering statistics. Valid read pairs (blue) represent successful Hi-C ligation products as described in (A). Self-fragment self-circles (light pink) are non-useful reads as they represent the same genomic fragment shown in (A). Same-fragment dangling ends (orange) represent reads in which a single restriction fragment was sequenced. Filtered and dumped pairs (brown) are also non-useful reads that have the wrong size or for which the ligation product could not be reconstructed. Finally, re-ligation reads (red) represent reads in which two adjacent fragments were re-ligated, thus producing non-useful information. (E) Valid read pairs contact distribution in the genome. Unique cis contacts (blue) are more frequent than unique trans contacts (green). Please click here to view a larger version of this figure.
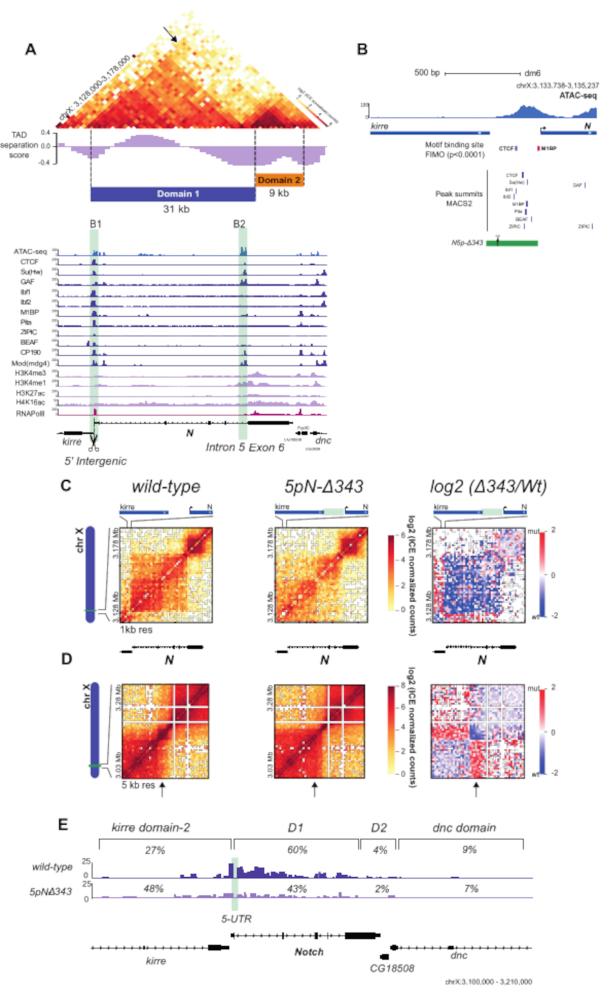
Figure 4: Hi-C contact matrices and virtual 4C analysis of WT and mutant S2R+ cells. (A) Hi-C normalized heatmap of a 50 kb region at 1-kb resolution centered in the Notch gene locus. TAD separation score32 for the locus is shown, along with the partitioning of the Notch locus into two topological domains (Domain 1 and Domain 2). ChIP-seq data for APs, RNA Pol II, and histone marks for S2/S2R+ cells34,35,36,37 are shown below the heatmap (Table 1). The positions of the Notch domain 1 boundaries are highlighted in light green. (B) Schematic representation of B1 boundary CRISPR mutant. The green rectangle indicates the deleted 343 bp region. Scissors indicate sgRNAs used for CRISPR-mediated genome editing. Motif-binding sites for APs are shown as boxes for CTCF and M1BP. Peak summits for DNA-binding APs shown in (A) are also indicated35. (C) Hi-C normalized heatmaps at 1 kb resolution covering a 50 kb region centered in Notch for the WT and the mutant cells. Left, Hi-C heatmaps of the log2 differences in interaction frequency between WT and mutant cells. (D) Hi-C normalized heatmaps covering a 250 kb region centered in Notch at 5 kb resolution for WT and mutant cells. Left, Hi-C heatmaps of the log2 differences in interaction frequency between WT and mutant cells. (E) Virtual-4C for WT and mutant cells using the 5' UTR of Notch as viewpoint. The percentages of interactions between the viewpoint and regions within the upstream kirredomain-2, Notch domain 1, Notch domain 2, and the downstream dnc domain for both WT and mutant cells are shown25. Abbreviations: WT = wild type; TAD = topologically associated domain; ChIP = chromatin immunoprecipitation; AP = architectural protein; RNA Pol = RNA polymerase; S2R+ = S2 receptor plus; sg RNA = single guide RNA; UTR = untranslated region. Please click here to view a larger version of this figure.
| Experiment | Sample | GEO Accession number |
| ChIP | CP190 | GSM1015404 |
| ChIP | SuHW | GSM1015406 |
| ChIP | Mod(mdg4) | GSM1015408 |
| ChIP | CTCF | GSM1015410 |
| ChIP | Ibf1 | GSM1133264 |
| ChIP | Ibf2 | GSM1133265 |
| ChIP | BEAF32 | GSM1278639 |
| ChIP | Pita | GSM1313420 |
| ChIP | ZIPIC | GSM1313421 |
| ChIP | RNA PolII | GSM2259975 |
| ChIP | H3K4me1 | GSM2259983 |
| ChIP | H3K4me3 | GSM2259985 |
| ChIP | H3K27ac | GSM2259987 |
| ChIP | MSL2 | GSM2469507 |
| ChIP | H4K16ac | GSM2469508 |
| ChIP | M1BP | GSM2706055 |
| ChIP | GAF | GSM2860390 |
| ChIP | Input | GSM1015412 |
| Hi-C | S2R+ WT cells | GSE136137 |
| Hi-C | S2R+ 5pN-delta343 cells | GSE136137 |
Table 1: GEO accession numbers.
| HiC-Pro statistics | Reads | Percentage |
| Mapping Statistics | ||
| Full read Alignments | 131515921 | 82.20% |
| Trimmed read Alignments | 16408309 | 10.30% |
| Failed to align | 12110964 | 7.60% |
| Pairing Statistics | ||
| Uniquely Aligned | 79428455 | 50.90% |
| Singleton | 19063418 | 12.20% |
| Multi Aligned | 57700021 | 36.90% |
| Filtering Statistics | ||
| Valid Pairs | 71373989 | 90.12% |
| Same-Fragment: Self-circle | 2340697 | 2.90% |
| Same-Fragment: Dangling Ends | 2578783 | 3.20% |
| Filtered Pairs | 2773043 | 3.50% |
| Dumped pairs | 196565 | 0.20% |
| Contact Statistics | ||
| Unique: cis ≤ 20 kbp | 33108815 | 46.50% |
| Unique: cis > 20 kbp | 33539888 | 47.10% |
| Unique: trans | 4133602 | 5.80% |
| Duplicate read pairs | 398400 | 0.60% |
Table 2: HiC-Pro statistics.
