In Vitro Differentiation of Naive CD4+ T Cells into Pathogenic Th17 Cells in Mouse
Summary
This protocol describes the differentiation of naïve CD4+ T cells into pathogenic Th17 cells in vitro. Specifically, when combined with a multi-parameter flow cytometry-based approach, 90% purity of pathogenic Th17 cells can be obtained from naïve CD4+ T cells using this differentiation method.
Abstract
In vitro T cell differentiation techniques are essential for both functional and mechanistic investigations of CD4+ T cells. Pathogenic Th17 cells have been linked to a wide range of diseases in recent times, including multiple sclerosis (MS), rheumatoid arthritis, acute respiratory distress syndrome (ARDS), sepsis, and other autoimmune disorders. However, the currently known in vitro differentiation protocols have difficulty achieving high purity of pathogenic Th17 cells, with the induction efficiency often below 50%, which is a key challenge in in vitro experiments. In this protocol, we propose an enhanced in vitro culture and differentiation protocol for pathogenic Th17 cells, which is used to directly differentiate naive CD4+ T cells isolated from mouse spleens into pathogenic Th17 cells. This protocol provides detailed instructions on splenocyte isolation, purification of naive CD4+ T cells, and differentiation of pathogenic Th17 cells. Through this protocol, we can achieve a differentiation purity of approximately 90% for pathogenic Th17 cells, which meets the basic needs of many cellular experiments.
Introduction
After leaving the thymus, naive CD4+ T lymphocytes pass through secondary lymphoid organs. Antigen-presenting cells that transmit homologous antigens to naïve CD4+ T cells activate them, starting a series of differentiation programs that eventually result in the production of highly specialized T helper (Th) cell lineages1. Interleukin 17 (IL-17) production characterizes Th17 cells, a subpopulation of pro-inflammatory Th cells2. Th17 cells play a role in the host's defense against extracellular pathogens and in the pathogenesis of many autoimmune diseases, such as autoimmune uveitis and multiple sclerosis. Signals from T-cell receptors and cytokines IL-6 and transforming growth factor-β (TGF-β) induce the differentiation of naive T cells into Th17 cells through the phosphorylation of signal transducer and activator of transcription (STAT)33. STAT3 is further amplified in a positive feedback loop through signaling mediated by IL-23 and IL-214,5. Phosphorylation of STAT3 can induce the expression of the transcription factors RORγt and RORα, which act as master switches regulating the cytokine profile of IL-17A, IL-17F, IL-21, and IL-22 in Th17 cells6. However, it has been reported that IL-6- and TGF-β-induced Th17 cells are insufficient to trigger autoimmune diseases, which require co-stimulation by IL-23 or separate co-stimulation of IL-6, IL-1β, and IL-23 in the absence of TGF-β7,8.
Th17 subsets that cannot effectively induce experimental autoimmune encephalomyelitis (EAE) are sometimes referred to as non-pathogenic Th17 while Th17 subsets that can induce EAE are known as pathogenic Th179. Current studies have shown that although pathogenic Th17 and non-pathogenic Th17 co-express the core transcription factor RORγt, there are great differences in the ability to produce IL-17A and pro-inflammatory and anti-inflammatory properties10. In addition to the high expression of RORγt, CCR6, STAT4, and RUNX4, which are common characteristic transcription factors of Th17, pathogenic Th17 cells also show additional gene signal expression characteristics related to the disease, such as TBX21, IFN-γ, and CXCR3, which have the characteristics of Th1 cell subsets. Pathogenic Th17 cells can secrete high levels of granulocyte-macrophage colony-stimulating factor (GM-CSF), IFN-γ, TNF-α, and other cytokines11,12. The phenotype of non-pathogenic Th17 cells is unstable, and only under the stimulation of CD3 and cytokine IL-2 can some of these cells differentiate into pathogenic Th17 cells. Therefore, in common clinical disease models such as rheumatoid arthritis, multiple sclerosis, and acute respiratory distress syndrome, pathogenic Th17 cells primarily exert pathogenic effects.
Pathogenic and non-pathogenic Th17 cells can differentiate in vitro under the influence of different cytokines. In recent years, several studies have proposed methods for inducing the differentiation of Th17 cells using different types and concentrations of cytokines. Th17 cells are stimulated by a combination of IL-6, IL-1β, and IL-2313,14,15,16. It has been proved that IL-6 and TGF-β, two cytokines necessary for Th17 cell differentiation, synergistically regulate the expression of RORγt and Th17 cell differentiation by interacting with two different conserved non-coding DNA sequences at the Rorc gene locus17. The stable phase of pathogenic Th17 cells is mainly maintained by IL-2318,19. IL-23 binds to its receptor and activates the JAK-STAT signaling pathway20, thereby causing phosphorylation of Jak2 and Tyk2 and promoting phosphorylation of STAT1, STAT3, STAT4, and STAT5. IL-4 and IFN-γ are negative regulators of this pathway. However, studies have shown that IL-1β may positively regulate the transcription of Rorα and Rorγt through the mTOR pathway to maintain the stability of the Th17 cell phenotype21.
Due to the heterogeneity of numerous studies, we chose the induction protocols for pathogenic and non-pathogenic Th17 cells from the latest research as controls22. The results indicate that, assuming that everything is carried out according to this protocol, after 5 days of culture under the condition of generating pathogenic Th17, more than 90% of the surviving cells can be pathogenic Th17 cells.
Protocol
The Institutional Review Committee for Animal Studies at Southeast University approved all the animal studies that are detailed in this study, which were carried out in compliance with both local and institutional office standards. Spleen samples were taken from C57BL6/J mice. Both female and male mice, aged between 5 and 8 weeks, were included in this study. The culture medium and buffer were stored at 4 °C for up to 1 month. Surgical instruments were autoclaved before use. Wear latex gloves and masks to avoid contamination of skin, eyes, and clothing with reagents; use a lot of water or saline rinse for the skin and eyes.
1. Precoating the 24-well tissue culture plate with anti-CD3
- Dilute anti-CD3 to a final concentration of 1 µg/mL in sterile 1x phosphate-buffered saline (PBS) or RPMI 1640 medium.
- Fill a 24-well tissue culture plate well with 1 mL of anti-CD3 solution (1 µg/mL); then, cover the well with parafilm.
- Place the anti-CD3 coated plate in the refrigerator (2 °C-8 °C) for 16 h or keep it at 37 °C in a cell incubator for 2-3 h.
2. Mouse spleen isolation and preparation of spleen single-cell suspension
- Induce complete anesthesia in C57BL/6J with 3% isoflurane and euthanize by inhaling 100% carbon dioxide for 2 min. Immediately after the mice's death, soak them in 75% ethanol for disinfection to prevent contamination.
- Place the mouse in a supine position on the clean bench, cut it open along the midline of the abdomen, and separate the abdominal structures layer by layer. Gently open the greater omentum and stomach with tweezers, gently pull out the spleen with the gastrosplenic ligament, and bluntly separate the spleen from surrounding tissues and ligaments to obtain an intact spleen (be careful to avoid spleen crushing or rupture).
NOTE: To maintain a sterile state, the following operation is carried out in a sterile super clean bench. - Place a 70 µm cell filter in a 100 mm sterile culture dish, add the spleen to the cell strainer, and crush it with a syringe plunger. At the same time, add 5-8 mL of homogenate flushing fluid to push all the cells through the filter into the culture dish.
NOTE: The composition of the lymphocyte separation liquid kit can be seen in the Table of Materials. - Centrifuge the filtrate at 450 × g for 5 min at room temperature. Discard the supernatant.
- Resuspend the cell pellet with the sample diluent provided with the lymphocyte separation kit or with PBS or RPMI 1640 medium. Adjust the cell concentration of the cell suspension to 2 × 108-1 × 109 cells/mL.
NOTE: Typically, 4-6 mL of sample diluent is required for one spleen. - In a centrifuge tube, take the same amount of lymphocyte separation solution as the tissue single-cell suspension. Carefully pipette the single-cell suspension onto the surface of the lymphocyte separation solution and centrifuge at 800 × g, 25 °C for 30 min. Set the Acceleration and Deceleration of the centrifuge to an appropriate range (e.g., set at 3 if there are the usual 1 to 9 gears).
NOTE: The lymphocyte separation solution should not be less than 3 mL. - After centrifugation, observe the four layers in the centrifuge tube from the top to the bottom. The first layer corresponds to the sample diluent, followed by the annular milky white lymphocyte layer. The third layer is the separation liquid, and the fourth layer consists of red blood cells.
- Use a pipette to carefully draw the second layer of annular milky white lymphocyte layer into another centrifuge tube and add 10 mL of cleaning solution to the centrifuge tube to mix the cells.
- Centrifuge at 250 × g for 5 min at room temperature. Discard the supernatant. Use 10 mL of RPMI 1640 medium to resuspend the cell pellet for cell counting.
3. Purification of naïve CD4+ T cells based on negative selection of magnetic beads
NOTE: Isolate untouched and highly purified naïve CD4+ T cells (CD4+CD44lowCD62Lhigh) from mouse splenocytes by immunomagnetic negative selection.
- Prepare the sample to have 1 × 108 cells/mL within the volume range of 0.1-2 mL.
- Add 50 µL/mL of rat serum and transfer the sample to a 5 mL polystyrene round-bottom tube.
NOTE: The composition of the naive CD4 T cell sorting kit can be seen in the Table of Materials. - Add 50 µL/mL of Isolation Cocktail to the sample, mix well, and incubate for 7.5 min at room temperature.
- Add 50 µL/mL of Depletion Cocktail to the sample, mix well, and incubate for 2.5 min at room temperature.
- Vortex the magnetic beads to ensure even dispersion. Add 75 µL/mL of the magnetic beads to the sample, mix well, and incubate for 2.5 min at room temperature.
NOTE: The particles should appear evenly dispersed; vortexing time should not be less than 30 s. - Top up the sample with RPMI 1640 medium to a final volume of 2.5 mL. Mix several times by gently pipetting up and down.
- Place the tube (without the lid) into the magnet and incubate for 2.5 min at room temperature.
- Picking up the magnet, invert the magnet and tube in one continuous motion and pour theenriched cell suspension into a new tube.
NOTE: Keep the tube and magnet inverted for 2-3 s before turning them upright. Do not disturb any drops that may remain on the mouth of the tube. - Determine the cell number using a hemocytometer.
- Perform flow cytometric analysis for naïve CD4+ T cells before and after isolation (Figure 1).
- Transfer the cells to a new centrifuge tube, centrifuge at 400 × g for 5 min at room temperature, and discard the supernatant.
- Resuspend the cells in 200-500 µL of RPMI 1640 medium (supplemented with 10% fetal bovine serum). For each 1 mL of cell culture (e.g., ~106 cells/mL), add 2 µL of leukocyte activation cocktail and mix it. Culture the cells in a CO2 incubator at 37 °C and saturated humidity for 4-6 h.
NOTE: The cells were vortexed once every 1-2 h. - Centrifuge the cells at 400 × g, 4 °C for 5 min. Carefully discard the supernatant and resuspend the cells in 200 µL of PBS. Add 1 µL of Fc blocker to the cells (e.g., ~106 cells/mL). Incubate the cells for 10 min at 4 °C.
- Wash the cells once with 1 mL of PBS by centrifuging at 400 × g for 5 min at 4 °C. Discard the supernatant and resuspend the cells in 200 µL of PBS.
- Add antibody cocktail (Fixable Viability Dye, 1:1,000; anti-CD4, 1:200; anti-CD44, 1:200; anti-CD62L, 1:200) and incubate the cells for 15-20 min in the dark at 4 °C.
- Wash the cells with 2 x 1 mL of PBS and then, centrifuge at 400 × g for 5 min at 4 °C.
- Discard the supernatant, resuspend the cells in 250 µL of fixed buffer, and incubate them for 40-60 min in the dark at 4 °C.
NOTE: The fix buffer is provided with the Foxp3/transcription factor staining buffer, which is a 4x stock solution that must be diluted with permeabilization diluent. - Add 1 mL of 1x permeabilization buffer to wash the cells 2x by centrifuging the cells at 300 × g, 4 °C for 5 min.
- Discard the supernatant, resuspend the cells in 200 µL of permeabilization buffer, and add the antibody (Foxp3 [nuclear], 1:200) to the cells. After fixation and membrane rupture, incubate the cell-antibody mixtures in the dark for 40-60 min with shaking occasionally for short periods at 4 °C.
NOTE: The Permeabilization/Wash solution is a 10x stock solution that must be diluted before use with PBS.
4. Induction of pathogenic Th17 cells in vitro
- Remove sterile PBS from the precoated 24-well plates prior to use.
- Use Th17 cell culture medium (Table 1) and contrast medium for culture, including Th0 cell culture medium (Table 1), classical non-pathogenic Th17 culture medium (Table 1), and classical pathogenic Th17 culture medium (Table 1). Resuspend the enriched naive CD4 + T cells and dispense into different wells in the same 24-well plate (precoated with anti-CD3), adjusting the concentration to 4 × 105 cells/mL, with 1 mL of medium in each well.
- Add 1 µL/mL of anti-CD28 solution (final concentration: diluted with medium to 2 µg/mL) to each well. Culture the cells in a 5% CO2 incubator at 37 °C for 5 days.
- Replace the cell culture supernatant medium with culture medium (Th0, non-pathogenic Th17, classical pathogenic Th17, and objective Th17) by carefully pipetting to distribute the cells evenly, discard half of the medium containing the cells, and then add new medium equal to the discard volume on day 2. Repeat on day 4.
- Observe the cells under an optical microscope on day 5 (Figure 2). Collect the cell supernatants from each group and cryopreserve them at -80 °C.
5. Flow cytometric analysis for pathogenic Th17 and Th0 cell differentiation
- Harvest all cells (per well) after 5 days of initiation of culture. Perform steps 3.12-3.15.
- Add an antibody cocktail (Fixable Viability Dye, 1:1,000; anti-CD4, 1:200) and incubate the cells for 15-20 min in the dark at 4 °C.
- Perform steps 3.17-3.19.
- Discard the supernatant, resuspend the cells in 200 µL of permeabilization buffer, and add the antibody cocktail (IL-17A [intracellular], 1:200; RORγT [nuclear], 1:200) to the cells. After fixation and membrane rupture, incubate the cell-antibody mixtures in the dark for 40-60 min with shaking occasionally for short periods at 4 °C.
- Add 1 mL of 1x Permeabilization/Wash solution to wash the cells 2x, centrifuging the cells at 400 × g for 5 min at 4 °C; then, discard the supernatant.
- Examine the cells on a flow cytometer and analyze the data using the software (Figure 3).
- Turn on the flow cytometer, and turn on the self-starting cleaning and calibration process.
- Select the laser channel used in this experiment, and set up a blank tube, single-stained tubes, and sample tubes.
- Click on the blank tube option and test on the machine; adjust the voltage so that the cell event is, as far as possible, in the center of the collection box.
- Collect single-stained tube cells and perform spectral detection of the corresponding channels to confirm that the fluorescent antibody is added correctly.
- Click the unmix button and let the machine automatically adjust the spectral compensation. With other flow cytometry instruments, adjust the fluorescence compensation normally.
- Collect each sample tube cell, save the data format as FCS files, and export. Analyze and plot in the relevant flow cytometry data analysis software.
- Circle the main cell population and remove possible cell adhesions through the diagonal circle gates of FSC-H and FSC-A.
- Circle live cells according to the FVS negative circle area; next, circle the CD4+ cell population.
- Construct the cross gate with the horizontal axis as IL-17 A and the vertical axis as RORγT; the double positive selection in the Q2 area is the Th17 cell population.
6. Enzyme-linked immunosorbent assay (ELISA) for detection of IL-17A secretion induced by different media
- Thaw the frozen cell supernatants collected in Section 4.5.
- Add 100 µL of the diluted standard, blank, and sample into the designated wells. Use the sealer to cover the plate and incubate for 90 min at 37 °C.
NOTE: Add the solutions to the bottom of the micro-ELISA plate well without touching the inside wall or causing any foaming. - Decant the liquid from each well and immediately add 100 µL of Biotinylated Detection Antibody working solution. Use a new sealer to cover the plate and incubate for 1 h at 37 °C.
- Decant the solution and add 350 µL of wash buffer to each well. After 1 min, aspirate or decant the solution from each well and pat it dry against clean absorbent paper. Repeat this wash step 3x.
- Add 100 µL of the horseradish peroxidase Conjugate working solution to each well. Use a new sealer to cover the plate and incubate for 30 min at 37 °C.
- Decant the solution from each well, and repeat the wash process 5x as described in step 6.4.
- Add 90 µL of the Substrate Reagent to each well, and incubate for about 15 min at 37 °C with a new sealer, protecting the plate from light.
NOTE: The reaction time can be altered based on the actual color change but should not exceed 30 min. Let the microplate reader warm up for ~15 min before OD measurement. - Add 50 µL/well of Stop Solution in the same order as the substrate solution.
- Determine the optical density (OD value) of each well at once with a microplate reader set to 450 nm.
- Obtain the OD values for the standard samples and replicate wells of the samples, and subtract the OD values of the blank wells to obtain the corrected values. Then, perform linear fitting with concentration as the abscissa and OD value as the ordinate. Based on the fitted equation, calculate the IL-17A concentration values for the sample wells.
NOTE: After 4 days of differentiation, the content of IL-17A in the supernatant of each group is shown in Figure 4. The fitting method can refer to the Orgin21 software. The standard curve obtained from this experiment has an R2 value of 0.9946.
7. T-cell differentiation by signature gene expression assays via quantitative PCR (qPCR)
NOTE: To rule out the possibility of flow cytometry instability due to the effects of dyes and fixation/rupture, we detected the expression of characteristic genes by qPCR to elucidate the differentiation effect of pathogenic Th17 at the transcriptional level.
- Measure the number of cells at the end of T cell culture. Collect the cells in 1.5 mL centrifuge tubes and centrifuge at 400 × g for 5 min. Carefully discard all the supernatants.
- RNA extraction
NOTE: The extraction process must be carried out in a fume hood or an ultra-clean bench to avoid RNA enzyme contamination.- Add 500 µL of Lysis Buffer (e.g., to 1-2 × 106 cells) to the sample tube (step 7.1), immediately shake and mix until there is no cell mass, and let it stand for 1 min.
- Add the mixture to the adsorption column placed in a collection tube, centrifuge at 13,400 × g for 30 s, and discard the filtrate.
- Add the specified amount of absolute ethanol to the Washing Buffer bottle before using it for the first time. Add 500 µL of washing buffer to the adsorption column, centrifuge at 13,400 × g for 30 s, and discard the filtrate.
- Repeat the washing in step 7.2.3.
- Place the adsorption column RA into the empty collection tube and centrifuge at 13,400 × g for 2 min to remove the rinse solution.
NOTE: The rinse solution must be removed to prevent the residual ethanol in the rinse solution from inhibiting the downstream reaction. - Remove the adsorption column RA and place it in a clean RNase-free centrifuge tube. Add 50 µL of RNase-free H2O to the middle of the adsorption membrane, let it stand for 1 min at room temperature, and centrifuge at 9,600 × g for 1 min.
- Measure the purity and concentration of RNA.
- Synthesize the first strand of cDNA by following the kit manufacturer's instructions.
- Remove genomic DNA. Take 500 ng of the extracted template RNA in an RNase-free centrifuge tube; add RNase-free ddH2O and 4x gDNA mix to form a mixture. Blend gently with a pipette, and place in a 42 °C water bath for 2 min.
NOTE: The amount of the reaction mixture and the amount added are related to the RNA concentration; refer to the instructions in detail. The 20 µL reaction system was used in this experiment. - Directly add a 5x reverse transcription reaction system in the reaction tube of the previous step. Take 4 µL of the reverse transcription reaction system and 16 µL of the mixture from the previous step and gently mix with a pipette.
NOTE: The reverse transcription system includes all the required reverse transcriptase, which is a directly configured reverse transcription system. - Set the reverse transcription reaction: 37 °C 15 min, 85 °C, 5 s, and finally cool to 4 °C.
- Remove genomic DNA. Take 500 ng of the extracted template RNA in an RNase-free centrifuge tube; add RNase-free ddH2O and 4x gDNA mix to form a mixture. Blend gently with a pipette, and place in a 42 °C water bath for 2 min.
- Set up the PCR reaction following the manufacturer's instructions.
- Add 10 µL of 2x PCR reaction enzyme mixture in the qPCR tube, 0.4 µL of primer 1, 0.4 µL of primer 2, 0.4 µL of 50x of ROX Reference Dye 1, 2 µL of template cDNA, and 6.8 µL of ddH2O to prepare a 20 µL mixture.
- Perform PCR in the real-time fluorescence quantitative PCR instrument using the following settings: stage 1, 95 °C, 30 s, Rep x 1; stage 2, first 95 °C, 10 s, then 60 °C, 30 s, Rep x 40; stage 3, first 95 °C, 15 s, next 60 °C, 60 s, finally 95 °C, 15 s, Rep x 1.
NOTE: After 4 days of differentiation, the relative expression levels of Il17a, Il17f, Rora, and Il23r in naïve CD4+ T cells are shown in Figure 5.
Representative Results
Our protocol was developed based on earlier research about pathogenic Th17 cell differentiation. The first step of the experiment is to detect the purity of naïve CD4+ T cells isolated from the spleen by magnetic bead sorting, which is the basis for the success of our subsequent pathogenic Th17 cell differentiation. The purity of naïve CD4+ T cells was detected using surface markers CD62L23 and CD4424 while FOXP325 was used as a marker of Treg cells. We found that the content of Treg cells was significantly reduced after sorting, and the purity of naïve CD4+ T cells could reach at least 95% (Figure 1). To compare the differentiation of pathogenic Th17 cells, naïve CD4+ T cells were cultured with Th0 medium (Table 1) and Th17 differentiation medium (Table 1) for a total of 5 days. It was found that T cells showed cluster growth in both Th0 and Th17 media (Figure 2).
Next, the cells were fixed, permeabilized, and labeled with antibodies against several cytokines in differentiated CD4+ T cells based on flow cytometry. We examined IL17A26 and RORγT27 as the hallmark cytokines of Th17 cell differentiation and found that 90% of naïve CD4+ T cells successfully differentiated into pathogenic Th17 cells under the stimulation of new Th17 cell culture medium (Figure 3). Figure 5 shows the signature genes of Th17 cells detected by PCR, which proved that the pathogenic Th17 cells we obtained by differentiation were stably expressing.
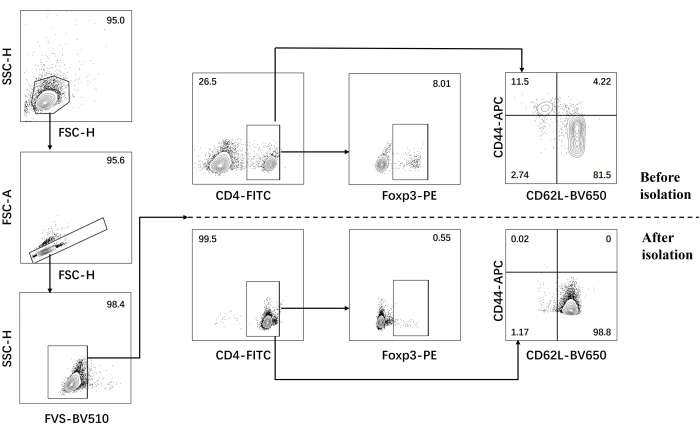
Figure 1: Gating strategy for analysis of signature cytokines in C57BL/6J mouse before and after naïve CD4+ T cell isolation. Abbreviations: FSC-H = forward scatter-peak height; SSC-H = side scatter-peah height; FSC-A = forward scatter-peak area; FITC = fluorescein isothiocyanate; PE = phycoerythrin. Please click here to view a larger version of this figure.
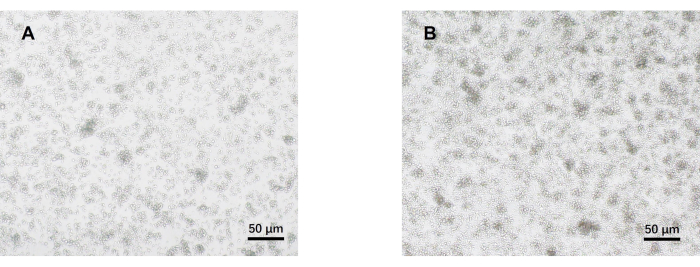
Figure 2: Representative images of mouse naïve CD4+ T cells cultured under pathogenic Th17 and Th0 conditions for 5 days. (A) Th0 cells; (B) pathogenic Th17 cells. Scale bars = 50 µm. Please click here to view a larger version of this figure.
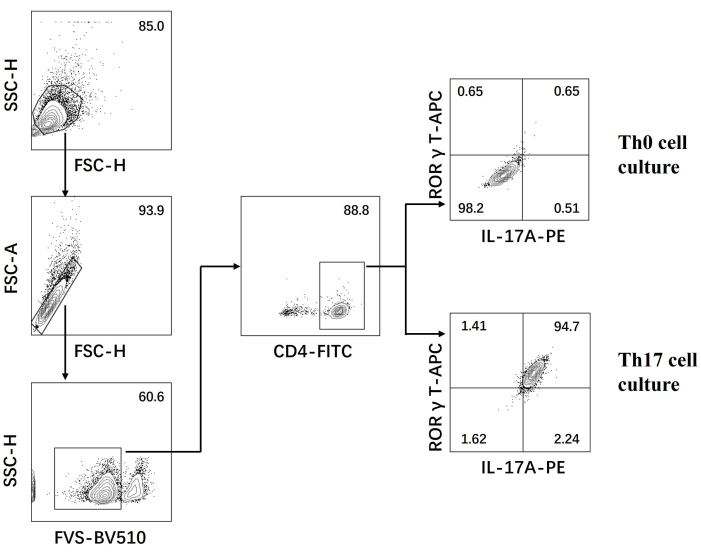
Figure 3: Flow cytometry analysis after differentiation induced by Th0 cell culture medium and Th17 cell culture medium. Abbreviations: FSC-H = forward scatter-peak height; SSC-H = side scatter-peah height; FSC-A = forward scatter-peak area; FITC = fluorescein isothiocyanate; PE = phycoerythrin. Please click here to view a larger version of this figure.
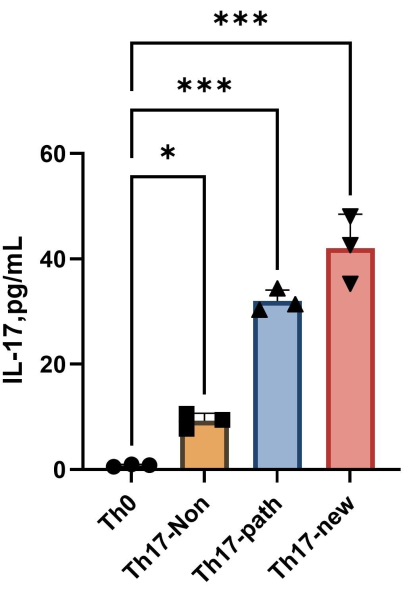
Figure 4: The content of IL-17A in the supernatant of Th0 cell culture medium and Th17 cell culture medium after induction of differentiation. Please click here to view a larger version of this figure.
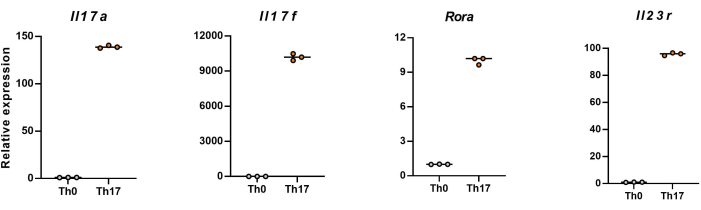
Figure 5: Representative results of the expression levels of signature cytokines in differentiated CD4+ T cells of C57BL/6J mouse. Please click here to view a larger version of this figure.
| Target pathogenic Th17 cell culture medium | Th0 cell culture medium | Classical non-pathogenic Th17 cell culture medium | Classical pathogenic Th17 cell culture medium. | |||||||||||
| Reagent | Final concentration | Amount | Final concentration | Amount | Final concentration | Amount | Final concentration | Amount | ||||||
| Penicillin-Streptomycin (100x) | 1x | 500 μL | 1x | 500 μL | 1x | 500 μL | 1x | 500 μL | ||||||
| Fetal Bovine Serum | 10% | 5 mL | 10% | 5 mL | 10% | 5 mL | 10% | 5 mL | ||||||
| β-mercaptoethanol | 50 μM | 50 μL | 50 μM | 50 μL | 50 μM | 50 μL | 50 μM | 50 μL | ||||||
| GlutaMAXTM supplement (100x) | 1x | 500 μL | 1x | 500 μL | 1x | 500 μL | 1x | 500 μL | ||||||
| Sodium pyruvate solution (100x) | 1 mM | 500 μL | 1 mM | 500 μL | 1 mM | 500 μL | 1 mM | 500 μL | ||||||
| Anti-Mouse IFN gamma (1 mg/mL) | 5 µg/mL | 250 μL | 10 µg/mL | 500 μL | 10 µg/mL | 500 μL | ||||||||
| Anti-Mouse IL-4 (2 mg/mL) | 5 µg/mL | 125 μL | 5 µg/mL | 250 μL | 10 µg/mL | 250 μL | ||||||||
| Mouse rIL-1 beta (20 µg) | 20 ng/mL | NA | ||||||||||||
| Mouse rIL-6 (20 µg) | 20 ng/mL | NA | 50 ng/mL | NA | 50 ng/mL | NA | ||||||||
| Mouse rIL-23 (50 µg) | 50 ng/mL | NA | 10 ng/mL | NA | ||||||||||
| Mouse TGF beta (100 µg) | 3 ng/mL | NA | 1 ng/mL | NA | 1 ng/mL | NA | ||||||||
| Murine IL-2 (5 µg) | 20 ng/mL | NA | ||||||||||||
| RPMI 1640 | NA | To 50 mL | NA | To 50 mL | NA | To 50 mL | ||||||||
| Total | NA | 50 mL | NA | 50 mL | NA | 50 mL | ||||||||
Table 1: Target pathogenic Th17 cell culture, Th0 cell culture, classical non-pathogenic and pathogenic Th17 cell culture media.
Discussion
This procedure offered a productive way to increase the number of mice splenic naïve CD4+ T cells for the in vitro production of pathogenic Th17 cells. Although we use more cytokines than other reported Th17 cell culture media, we are committed to optimizing the growth conditions of pathogenic Th17 cells. We are considering further optimization of our induced differentiation protocol.
Here, we simply used flow cytometry and qPCR to examine the production of hallmark cytokines. With a few minor modifications, this approach can also be used for other function tests, such as cell proliferation.
We used a Chinese-produced lymphocyte isolation kit to isolate mouse spleen lymphocytes because it is effective and time-saving. Lymphocyte separation solutions based on other brands can also achieve the purpose of separating mouse spleen lymphocytes through different steps. Another method is to directly lyse the red blood cells of the obtained spleen cell suspension; however, we found that spleen red blood cells often cannot be lysed at one time.
Some problems can arise during the execution of this protocol. First, the number of naïve CD4+ T cells obtained by magnetic bead sorting may be very low (protocol step 3). This could be attributed to the process of crushing organs being insufficient. It is important to ensure that the organ is properly homogenized. Increasing the frequency of rinsing during the homogenization process will improve the recovery rate. To obtain a higher number of splenic naïve CD4+ T cells, we suggest using younger mice (6-10 weeks old). There are various methods available for separating spleen lymphocytes, and the yield may vary depending on the separation liquid used. It is recommended to use a universally certified separation liquid and try to extract the lymphocyte layer as much as possible.
Second, the proportion of CD4+ T cells in flow cytometry may be <80% (protocol step 5). One possible cause of this issue could be an inaccurate splenocyte count, resulting in a cell count greater than that of the additional antibody cocktail and magnetic beads. To increase the effectiveness of naïve CD4+ T cell purification, cell counting must be precise. Additionally, 10% more antibody cocktail and magnetic beads can be used over what this protocol recommends. Lastly, flow cytometry can be performed immediately after sorting naïve CD4+ T cells.
Third, there may not be many T cell clusters formed during the culture, and most of the cells may have died during the differentiation of T cells (protocol step 4). The potential reason for this problem may be the inaccurate determination of cell numbers for naïve CD4+ T cells before seeding, resulting in a low cell density. It is recommended to adopt a more accurate counting method to achieve the desired cell density of approximately 4 × 105 cells/mL for each well in a 48-well plate. Another possible cause could be technical problems with the CO2 incubator, such as incorrect temperature or CO2 concentration. Lastly, excessive force while changing the cell culture medium could potentially cause cell death.
Fourth, the relative expression levels of the signature cytokine genes may be low (protocol step 7). To ensure the authenticity of the extracted RNA, it is recommended to use a nanodroplet detection concentration of more than 100 ng/mL. The potential reason for the decrease in concentration may be the unhealthy nature of cultured cells, such as a large part of the collected cells are dead or in the process of death. To obtain the true RNA concentration, it is necessary to resolve the situation that may lead to poor growth during cell culture. An alternative reason behind this concern might be the excessively low final cell count achieved during RNA extraction, possibly due to inadvertent cell loss during the supernatant disposal. Employing advanced RNA extraction techniques such as one-step RNA extraction kits could prove advantageous. The ideal OD260/OD280 ratio, as measured by Nanodrop, should fall within the range of 1.9-2.1. In the event of an excessively low ratio, protein contamination becomes a possibility. Increasing the frequency of RNase-free buffer washing may aid in mitigating this issue. Conversely, an uncharacteristically low ratio implies potential RNA degradation. To counteract this issue, it is recommended to employ RNase-free water and utilize 1.5 mL tubes for RNase decontamination purposes.
In conclusion, the current protocol describes the use of new cell culture medium to directly induce naive CD4+ T cells to differentiate into pathogenic Th17 cells in vitro. Compared with direct separation, there is no doubt that this method is more direct, inexpensive, and more efficient. The configuration of the medium is also very simple so that the constructed Th17 cells can be more intuitively used for subsequent experiments, providing a very good cell model for the study of many diseases.
Divulgations
The authors have nothing to disclose.
Acknowledgements
The work was supported by the National Key R&D Program of China (No.2022YFC2304604), National Natural Science Foundation of China (No.81971812), National Natural Science Foundation of China (No.82272235), Science Foundation of the Commission of Health of Jiangsu Province (No. ZDB2020009), Jiangsu Province Key research, development Program (Social Development) Special Project (No.BE2021734), National Key R & D Program of Ministry of Science and Technology (No.2020YFC083700), Jiangsu Provincial Key Laboratory of Critical Care Medicine (BM2020004), Key project of National Natural Science Foundation of China (81930058), National Natural Science Foundation of China (82171717), Central Universities Basic Research Funds (2242022K4007), and Jiangsu Provincial Natural Science Foundation General Project (BK20211170).
Materials
| Antibodies | |||
| Rat anti-mouse CD62L, BV650, clone MEL-14, 1:200 dilution | BD | Cat# 564108; RRID: AB_2738597 | |
| Rat monoclonal anti-CD4, FITC, clone RM4-5, 1:200 dilution | BioLegend | Cat#100509; RRID: AB_312712 | |
| Rat monoclonal anti-IL-17A, PE, clone TC11-18H10.1, 1:200 dilution | BioLegend | Cat#506903; RRID: AB_ 315463 | |
| Rat monoclonal anti mouse/human CD44, APC, clone IM&, 1:200 dilution | BioLegend | Cat#103012; RRID: AB_312963 | |
| Rat monoclonal anti-RORγT, APC, clone B2D, 1:200 dilution | Invitrogen | Cat#17-6981-80; RRID: AB_2573253 | |
| Rat monoclonal FOXP3 antibody, PE, clone FJK-16s, 1:200 dilution | Invitrogen | Cat#12-5773-82; RRID: AB_465936 | |
| Chemicals, peptides, and recombinant proteins | |||
| Anti-Mouse CD3 SAFIRE purified | biogems | Cat#05112-25 | |
| Anti-Mouse CD28 SAFIRE purified | biogems | Cat#10312-25 | |
| Anti-Mouse IFN gamma | biogems | Cat#80822-25 | |
| Anti-Mouse IL-4 | biogems | Cat#81112-25 | |
| Ethanol | Xilong scientific | Cat#64-17-5 | |
| Fetal bovine serum | Gibco | Cat#10437-028 | |
| FcR Blocking reagent | Miltenyi Biotec | Cat#130-092-575 | |
| GlutaMAX supplement | gibco | Cat#35050079 | |
| Mouse rIL-1 beta | Sino Biological | Cat#50101-MNAE | |
| Mouse rIL-6 | Sino Biological | Cat#50136-MNAE | |
| Mouse rIL-23 | Sino Biological | Cat#CT028-M08H | |
| Mouse TGF beta 1 | Sino Biological | Cat#50698-M08H | |
| PBS | Procell | Cat#PB180327 | |
| Recombinant Murine IL-2 | peprotech | Cat#212-12 | |
| RPMI 1640 with L-glutamine | Gibco | Cat#11875-119 | |
| Penicillin-streptomycin solution | Gibco | Cat#15070063 | |
| β-mercaptoethanol | Sigma-Aldrich | Cat#M6250-100ML | |
| Critical commercial assays | |||
| Animal Organ Lymphocyte Separation Solution Kit | Tbdscience | Cat#TBD0018SOP | Contains animal spleen tissue lymphocyte separation liquid, tissue sample diluent (cat no. 2010C1119), sample cleaning solution (cat no. 2010X1118), sample washing solution (cat no. TBTDM-W), tissue homogenate flushing liquid (F2013TBD) |
| ChamQ SYBR qPCR Master Mix (High ROX Premixed) | Vazyme | Cat#Q341-02 | https://www.vazymebiotech.com/product-center/chamq-sybr-qpcr-master-mix-high-rox-premixed-q341.html. |
| Fixation/permeabilization Concentrate | invitrogen | Cat#00-5123-43 | |
| Fixation / Permeabilization Diluent | invitrogen | Cat#00-5223-56 | |
| Fixable Viability Dye EF506 | invitrogen | Cat#65-0866 | |
| HiScript II Q RT SuperMix for qPCR (+gDNA wiper) | Vazyme | Cat#R223-01 | https://www.vazymebiotech.com/product-center/hiscript-ii-q-rt-supermix-for-qpcr-gdna-wiper-r223.html. |
| Leukocyte Activation Cocktail | BD | Cat#550583 | |
| Mouse IL-17A (Interleukin 17A) ELISA Kit | Elabscience® | Cat#E-EL-M0047 | |
| Naïve CD4+ T cells isolation kit, mouse | STEMCELL | Cat#19765 | EasySep kit contains mouse CD4+ T cell isolation cocktail [cat no. 19852C.1], mouse memory T cell depletion cocktail [cat no. 18766C], streptavidin RapidSphered 50001 [cat no. 50001], normal rat serum [cat no. 13551]); only store rat serum at -20 °C; other components to be stored at 2-8 °C. |
| Permeabilization Buffer | invitrogen | Cat#00-8333-56 | |
| SPARKeasy Cell RNA Kit | Sparkjade | Cat#AC0205-B | https://www.sparkjade.com/product/detail?id=85 |
| Experimental models: Organisms/strains | |||
| Mouse: C57BL/6 | Gempharmatech | Cat#000013 | |
| Oligonucleotides | |||
| Mouse Il17a TaqMan primers with probe | ribobio | NA | |
| Mouse Il17f TaqMan primers with probe | ribobio | NA | |
| Mouse Il23r TaqMan primers with probe | ribobio | NA | |
| Mouse Rora TaqMan primers with probe | ribobio | NA | |
| β-actin TaqMan primers with probe | ribobio | NA | |
| Software and algorithms | |||
| Cytek Aurora | Cytek | https://spectrum.cytekbio.com/ | |
| FlowJo v10.8.1 | Tree Star | https://www.flowjo.com | |
| GraphPad prism 9 | GraphPad Software | https://www.graphpad-prism.cn | |
| Other | |||
| 1 mL syringe | Kindly | NA | |
| 1.5 mL Centrifuge tubes | Eppendorf | Cat#MCT-150-C | |
| 5 mL Round-bottom tubes | Corning | Cat#352235 | |
| 15 mL Centrifuge tubes | NEST | Cat#601052 | |
| 48-Well tissue culture plate (flatten bottom) | Corning | Cat#3548 | |
| 50 mL Centrifuge tubes | NEST | Cat#602052 | |
| 70 µm Cell strainer | Biosharp | Cat#BS-70-XBS | |
| 96-well Unskirted qPCR Plates | VIOX scientific | Cat#V4801-M | |
| 100 mm Petri dish | Corning | Cat#430167 | |
| Centrifuge | Eppendorf | 5425R | |
| Cell culture CO2 incubator | Thermo Fisher | HEPA Class100 | |
| Cytek Aurora | Cytek | NA | |
| dissecting scissors | RWD | S12003-09 | |
| Hemocytometer | AlphaCell | Cat#J633201 | |
| NanoDrop 2000 Spectrophotometer | Thermo Fisher | ND-2000 | |
| Real-time PCR System | Roche | LightCycler96 | |
| Surgical tweezers | RWD | F12005-10 | |
| Thermal cycler | Bio-Rad | C1000 Touch |
References
- Korn, T., Bettelli, E., Oukka, M., Kuchroo, V. K. IL-17 and Th17 cells. Annu Rev Immunol. 27, 485-517 (2009).
- Bhaumik, S., Basu, R. Cellular and molecular dynamics of Th17 differentiation and its developmental plasticity in the intestinal immune response. Front Immunol. 8, 254 (2017).
- Bettelli, E., Korn, T., Oukka, M., Kuchroo, V. K. Induction and effector functions of T(H)17 cells. Nature. 453 (7198), 1051-1057 (2008).
- Korn, T., et al. IL-21 initiates an alternative pathway to induce proinflammatory T(H)17 cells. Nature. 448 (7152), 484-487 (2007).
- Bettelli, E., et al. Reciprocal developmental pathways for the generation of pathogenic effector TH17 and regulatory T cells. Nature. 441 (7090), 235-238 (2006).
- Yang, X. O., et al. T helper 17 lineage differentiation is programmed by orphan nuclear receptors ROR alpha and ROR gamma. Immunity. 28 (1), 29-39 (2008).
- Ghoreschi, K., et al. Generation of pathogenic T(H)17 cells in the absence of TGF-β signalling. Nature. 467 (7318), 967-971 (2010).
- Lee, Y., et al. Induction and molecular signature of pathogenic TH17 cells. Nat Immunol. 13 (10), 991-999 (2012).
- McGeachy, M. J., et al. The interleukin 23 receptor is essential for the terminal differentiation of interleukin 17-producing effector T helper cells in vivo. Nat Immunol. 10 (3), 314-324 (2009).
- Wu, B., et al. The TGF-β superfamily cytokine Activin-A is induced during autoimmune neuroinflammation and drives pathogenic Th17 cell differentiation. Immunity. 54 (2), 308-323 (2021).
- Ramesh, R., et al. Pro-inflammatory human Th17 cells selectively express P-glycoprotein and are refractory to glucocorticoids. J Exp Med. 211 (1), 89-104 (2014).
- Basdeo, S. A., et al. Ex-Th17 (nonclassical Th1) cells are functionally distinct from classical Th1 and Th17 cells and are not constrained by regulatory T cells. J Immunol. 198 (6), 2249-2259 (2017).
- Cua, D. J., et al. Interleukin-23 rather than interleukin-12 is the critical cytokine for autoimmune inflammation of the brain. Nature. 421 (6924), 744-748 (2003).
- Awasthi, A., et al. Cutting edge: IL-23 receptor gfp reporter mice reveal distinct populations of IL-17-producing cells. J Immunol. 182 (10), 5904-5908 (2009).
- Jäger, A., Dardalhon, V., Sobel, R. A., Bettelli, E., Kuchroo, V. K. Th1, Th17, and Th9 effector cells induce experimental autoimmune encephalomyelitis with different pathological phenotypes. J Immunol. 183 (11), 7169-7177 (2009).
- Lee, Y., Collins, M., Kuchroo, V. K. Unexpected targets and triggers of autoimmunity. J Clin Immunol. 34, S56-S60 (2014).
- Chang, D., et al. The conserved non-coding aequences CNS6 and CNS9 control cytokine-induced Rorc transcription during T helper 17 cell differentiation. Immunity. 53 (3), 614-626 (2020).
- Bunte, K., Beikler, T. Th17 cells and the IL-23/IL-17 axis in the pathogenesis of periodontitis and immune-mediated inflammatory diseases. Int J Mol Sci. 20 (14), 3394 (2019).
- Zhao, Y., Liu, Z., Qin, L., Wang, T., Bai, O. Insights into the mechanisms of Th17 differentiation and the Yin-Yang of Th17 cells in human diseases. Mol Immunol. 134, 109-117 (2021).
- Berghmans, N., et al. Interferon-γ orchestrates the number and function of Th17 cells in experimental autoimmune encephalomyelitis. J Interferon Cytokine Res. 31 (7), 575-587 (2011).
- Wu, B., Wan, Y. Molecular control of pathogenic Th17 cells in autoimmune diseases. Int Immunopharmacol. 80, 106187 (2020).
- Du, L., et al. Growth hormone releasing hormone signaling promotes Th17 cell differentiation and autoimmune inflammation. Nat Commun. 14 (1), 3298 (2023).
- Ernst, D. N., Weigle, W. O., Noonan, D. J., McQuitty, D. N., Hobbs, M. V. The age-associated increase in IFN-gamma synthesis by mouse CD8+ T cells correlates with shifts in the frequencies of cell subsets defined by membrane CD44, CD45RB, 3G11, and MEL-14 expression. J Immunol. 151 (2), 575-587 (1993).
- Radtke, A. J., et al. IBEX: an iterative immunolabeling and chemical bleaching method for high-content imaging of diverse tissues. Nat Protoc. 17 (2), 378-401 (2022).
- El-Hindi, K., et al. T-cell-specific CerS4 depletion prolonged inflammation and enhanced tumor burden in the AOM/DSS-induced CAC model. Int J Mol Sci. 23 (3), 1866 (2022).
- Cooney, L. A., Towery, K., Endres, J., Fox, D. A. Sensitivity and resistance to regulation by IL-4 during Th17 maturation. J Immunol. 187 (9), 4440-4450 (2011).
- Zhu, X., et al. A novel interleukin-2-based fusion molecule, HCW9302, differentially promotes regulatory T cell expansion to treat atherosclerosis in mice. Front Immunol. 14, 1114802 (2023).

.