1. Preparation of MOF suspensions using a polymer stabilizer
- Weigh out 50 mg of bis-amino-terminated polyethylene glycol (PNH2, Mn ~1,500) (see Table of Materials) and transfer to a one-dram vial (Table of Materials). Weigh out 1-5 mg of PCN-222(fb) (see synthetic protocol11) and place it into the same vial with PNH2.
NOTE: To attain the best possible MOF suspensions, the synthetic conditions required to make the MOF particle sizes need to be at or below 1 µm. - Find a suitable solvent (if not water, use an anhydrous solvent such as dimethylformamide [DMF] or Acetonitrile [ACN]; see Table of Materials) to suspend the MOF, making sure that the solvent window is wide enough so that the solvent itself is not excited with the wavelength of choice. Transfer 1-3 mL of solvent to the vial using an autopipette fitted with an appropriate pipette tip.
NOTE: The commonly used solvents noted above have wide solvent windows-CH3CN: 200 nm high-energy cut-off; and DMF: 270 nm cut-off. Solvents with higher refractive indices (1.4-1.5), like DMF, DMSO, and toluene, can be used to help minimize light scatter by being more closely aligned with the refractive index of quartz glass (ca. 1.46-1.55), thereby minimizing the bending of light in unwanted directions when passing through the cuvette. - Using a tip sonicator (see Table of Materials), sonicate the vial contents for 2-5 min at 20%-30% amplitude (i.e., the distance in which the sonicator tip longitudinally moves, typically ~60 µm for a 3 mm diameter probe at 30% amplitude) with intervals of 2 s on and 2 s off. This procedure serves to break up MOF aggregates and helps coat MOF particles with polymer. Ensure that the MOF suspension is well-dispersed and homogeneous by the end of the sonication process.
NOTE: Sonication times vary depending on how well the MOF inherently disperses. - Open up a fresh 10 mL plastic syringe (Table of Materials) and draw up the suspension into the syringe. Remove the syringe needle and replace it with a polytetrafluoroethylene (PTFE)-mesh 200 nm syringe filter (Table of Materials). Pass the suspension through the syringe filter into a new clean vial. The resulting suspension is ready for transient absorption spectroscopic measurements.
NOTE: Given that the average particle sizes of some MOFs exceed 200 nm, choosing the appropriate size is up to the user's discretion.
2. Preparation of filtered MOF suspensions for nanosecond transient absorption measurements (nsTA)
- With the filtered MOF suspension obtained in section 1, the absorption spectrum of the suspension needs to be obtained (Table of Materials). Wash a cuvette (1 cm path length) that is capable of sealing and purging (Table of Materials) with solvent three to five times, then fill it with 3 mL of DMF.
- With an absorption spectrophotometer, choose a wavelength region to measure the solvent and suspension. Measure the blank ultraviolet-visible (UV-Vis) spectrum of the solvent in the cuvette and set it as a background scan to be subtracted from the sample scans. Empty the solvent contents of the cuvette and transfer the MOF suspension into the cuvette.
NOTE: The electronic absorption spectrum of the PNH2 stabilizer (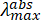 ~250 nm) has a weak absorption tail that persists until 450 nm, with an absorbance of ~0.01 at 450 nm at the initial concentration.
~250 nm) has a weak absorption tail that persists until 450 nm, with an absorbance of ~0.01 at 450 nm at the initial concentration. - Measure the absorption spectrum of the initial MOF suspension, keeping in mind the desired wavelength needed to excite the MOF sample. If the MOF suspension has an absorbance or optical density (OD) >1 at the desired excitation wavelength, then dilute with solvent and measure the absorbance spectrum until the OD is ≤1 at the excitation wavelength.
NOTE: For narrow-angle transient absorption measurements, repeat the absorption measurements to attain an appropriate OD at the excitation wavelength using a 2 mm cell (Table of Materials). For nanosecond transient absorption measurements of MOFs, an absorbance or OD of 0.1-1 at the excitation wavelength is needed to follow Beer's Law. The required OD is a wide range because some samples absorb strongly in different regions. A perfect example of this is porphyrins. Porphyrins have a strong narrow Soret band transition between 400-450 nm, whereas their Q-band transitions between 500-800 nm are quite weak. If one wanted to excite at one of their Q-bands, prepping a solution with an OD ~0.5 at one of the Q-bands would consequently exhibit a Soret band absorption >3, and the transient absorption detector would not be able to quantitatively process the changes in this region. Ultimately, it is at the user's discretion to determine the appropriate excitation wavelength and absorbance amplitude that allows for quantitative measurements in the desired spectral window.
3. Purging the MOF suspension
- With the MOF suspension adjusted to have the desired absorbance spectrum for TA measurements, place a 2 mm x 8 mm stir bar (Table of Materials) into the cuvette, and seal the inlet cuvette joint with a rubber septum.
- Take a 1 mL plastic syringe (Table of Materials), cut half of it with a pair of scissors, and keep the half of the syringe that allows for needles to be attached to it.
- With one end of a flexible tube attached to an Ar or N2 tank (Table of Materials), insert the syringe half into the other end of the tubing with the needle end outside.
- Wrap the stem of the exposed syringe half with parafilm (Table of Materials) to create a seal with the tube and syringe. If a hose clamp is available, it can be used instead of parafilm to create a seal with the syringe and purging tube.
- Attach a long purge needle (3 in, 25 G) (Table of Materials) to the syringe end and insert the needle into the sealed cuvette into the suspension. Take the needle from the 1 mL syringe (step 3.2) and insert it into the cuvette. Turn on the Ar or N2 flow and purge the suspension for 45 min-1 h.
NOTE: A technique known as "double-purging" is often used for solvents with a boiling point <100 °C. To employ this technique, a sealed flask with solvent is purged with the inlet needle, with one end of a cannula inserted into the flask headspace and the other end of the cannula inserted into the cuvette suspension. The outlet needle is inserted into the cuvette headspace. Purging this way minimizes solvent loss from evaporation over time. - After purging is finished, remove the needles and wrap the cuvette septum with four to five 2 in slices of parafilm (Table of Materials). Measure the absorption spectrum of the sample to make sure it matches the standards set in section 2. The sample is now ready for transient absorption measurements.
4. Perpendicular pump-probe nanosecond transient absorption setup (nsTA)
- Turn on the laser and nsTA spectrometer systems (Table of Materials; Figure 1). Adjust the laser output power to a low enough level, such that placing a white business card in the beam path allows for clear visibility of the laser spot, but not so bright that it is blinding.
- Open up both the mechanical laser shutter and the probe beam shutter so that they are both in the path of the sample holder.
- Adjust the vertical and horizontal position of the laser beam (Figure 1, P3), such that it hits the center of the sample cell holder (Figure 1, SC1) where the sample will be placed. Use a business card to confirm the position. Place a neutral density (ND) filter (OD 2; Table of Materials) in the path of the probe beam.
NOTE: All mirrors and prisms present in the nsTA system described herein are mounted on kinematic mounts (Table of Materials), and the beam positions are adjusted by manually turning the vertical and horizontal knobs on the mounts. A longer, 1 cm wide white card can be placed across the inside of an empty cuvette in SC1 to make for an easier alignment. - Place a cut business card (~1.5 cm width) in the sample holder (or hold it in the sample holder) and angle it across the sample holder such that both the laser beam and probe beam are hitting the same side of the business card. Fine-tune the laser beam position vertically (P3) to obtain the best overlap with the most intense part of the probe beam.
- Close the shutters, remove the ND filter, and place the sample in the sample chamber along with a magnetic stirrer (Table of Materials). TA measurements can now be taken.
- The system and software used in this work are provided in the Table of Materials. In the software suite, there are selection boxes titled Spectral Absorption and Kinetic Absorption for measuring TA spectra and absorption kinetics, respectively. Select the Spectral Absorption button and select the Setup mode.
- Set the time zero of the laser pulse in the software setup window by adjusting the input time in increments of +0.010 µs (e.g., -0.020 µs, -0.010 µs, 0.000 µs, 0.010 µs, etc.) until the laser pulse is not observed in the probe beam spectrum.
- With the time zero set, optimize the amount of light hitting the charge-coupled device (CCD) detector in the setup window by adjusting the bandwidth, gain, and gate width to reach a high enough count to obtain an adequate signal while not saturating the detector.
NOTE: We leave this process up to the user as detectors vary from system to system. - To collect a TA spectrum, click on the Multiple button in the Spectral Absorption tab. Make sure the settings from the setup window are present in this window. If the sample emits light, click on the Backgrounds tab and click on the Subtract Fluorescence Background button. For a cursory scan, set the averages to 4 to make sure a quality initial TA spectrum is obtained. If a satisfactory TA spectrum is obtained, obtain another one with more averages.
- To map a TA spectrum at different time delays after time zero, select the Map button in the Spectral Absorption tab. Ensure that the setup parameters have not changed in this tab. Enter the desired time intervals for mapping, click on Apply, and then click on Start to map the spectra.
- To obtain absorption kinetics at desired wavelengths, click on the Kinetic Absorption button in the software and click the Setup button in the drop-down menu. Enter the wavelength of interest in the Controller tab in the setup window and adjust the bandwidth to a suitable level. Usually, a bandwidth of 1 nm is a good starting point.
- In the Oscilloscope tab, adjust the photomultiplier tube (PMT) detector time window so that it is long enough to see the entire kinetic trace, from before laser excitation to the signal fully decaying back down to baseline. A usual starting point is a 4,000 ns window. Adjust the PMT voltage range to a suitable level, where the entire TA trace is observable on the signal axis. A voltage range of 160 mV is reasonable to begin measurements. Click on Apply and then Start. If the signal is too low or the time window is too short or too long, click on Stop and adjust the bandwidth and time window to suitable levels, making sure not to set the bandwidth too high to saturate/damage the detector.
- With the kinetic trace properly set up, close the Setup window and open the Multiple window from the drop-down menu after clicking on the Kinetic Absorption button. Check to make sure the parameters from the Setup window are the same in the Multiple window. Set the desired number of measurements (laser shots). Usually, 20 measurements are satisfactory for high-signal regions of the TA spectrum. If the sample emits at the probe wavelength, be sure to check the Subtract Fluorescence Background button in the Backgrounds tab. Click on Apply then Start to collect the TA kinetics.
NOTE: Sometimes, performing a higher number of shots (>40) shifts the baseline of the decay, positive or negative, from probe/laser scatter interference. If this is an issue, then perform a lower number of shots (~10-20) and repeat the measurement multiple times to collect multiple sets of data that can then be averaged together. - Once the TA measurements are complete, measure the absorption spectrum of the MOF afterward to ensure minimal degradation.
5. Narrow-angle nsTA setup
- Sometimes with the perpendicular pump-probe setups, the signal obtained from the MOF suspension is quite weak (<10 ΔmOD) and still fluctuates from scatter due to the large sample volume being excited. To help minimize the signal fluctuations and enhance the signal, ultrafast transient absorption setups can be applied toward nsTA setups with a narrow-angle pump-probe beam orientation and smaller path lengths (Figure 2).
- Depending on the sample chamber setup, one can focus and direct the excitation beam so that the pump and probe beams cross at angles <45°, and therefore provide more overlap. Do this with focusing optics (Figure 2, concave lens [CCL] and convex lens [CVL]) and kinematic mirrors (Figure 2, MM1-3). Turn on the laser/spectrometer system and repeat steps 4.2 and 4.3.
NOTE: While the use of concave/convex lenses is ideal for focusing optics, an optical iris can be used in place of these components to narrow the beam. The narrowing of the beam in this manner can be compensated for by increasing the power; however, when working with wavelengths below 400 nm, degradation and bleaching of the iris is quite common. Some TA spectrometers do not have breadboards that permit mounting optics in the sample chamber. The spectrometer used here does not have breadboards, so holes were drilled and tapped in the sample chamber to set up optics (Figure 2, MM1-3). If the spectrometer is still under warranty, contact the company support team to see if they can accommodate such a setup. - To decrease the beam spot size hitting the 2 mm cuvette (Table of Materials), set up a Galilean telescope with a concave lens (Table of Materials, CCL1) hitting the laser first and a convex lens (Table of Materials, CVL1) hitting the laser second. Ensure that the distance between the two lenses is approximately the difference between the two focal distances of the lenses.
NOTE: The Spectra-Physics Quanta Ray lasers used in these measurements have a spot size of 1 cm. The spot size was halved with the Galilean telescope setup. For lasers that output megawatts of power, Galilean telescopes need to be exclusively used. A Keplerian telescope (two convex lenses) forms plasma between the two lenses at even modest powers (~10 mW). - Open up both the laser and probe shutters. Replace the first shutter mirror (SM1) with SM2 and place a note card into the SM2 clamping mount, such that its orientation is completely facing the probe beam. Then, set up a series of mini mirrors (MM1-3), approximately as depicted in Figure 2. Direct the incoming laser beam by adjusting the turning knobs on the P3 kinematic mount approximately onto the center of MM1. To minimize laser beam expansion from mirror to mirror, place MM2 in front of MM1 to lower the angle of reflection between the two mirrors (Figure 2).
NOTE: For laser alignments, a common practice is to adjust a mirror/prism one mirror away from the intended spot location (e.g., adjusting P2 to precisely hit MM1). However, P2 in the experiments discussed here is a stationary beam-guiding optic and should not be adjusted. If given the flexibility, alignment should be done with an optical component one mirror away from the target optic. - With the beam hitting approximately the center on MM1, rotate MM1 so that the reflected laser beam is hitting MM2 in the center. With the beam hitting approximately the center on MM2, rotate MM2 so that the reflected laser beam is hitting MM3 in the center. With the beam hitting approximately the center on MM3, rotate MM3 so that the reflected laser beam is hitting the alignment note card in the same spot as the probe beam.
- Fine-tune the laser beam positions on each of the mirrors and note card with the vertical and horizontal knobs on the mirrors. Ensure that the beam has little to no clipping throughout its path.
- Repeat steps 5.5 and 5.6 using a 2 mm cuvette with a 14/20 inner joint (SC2) and 14/20 rubber septum (Table of Materials). Insert the sample into a clamping sample mount (SM2) completely facing the probe beam path. Fine-tune the laser beam positions on each mirror and SM2 with the vertical and horizontal knobs on the mirrors.
NOTE: For added ease in changing between perpendicular and narrow-angle TA setups, a flipping or magnetic kinematic mirror mount for MM1 can be used instead of a regular kinematic mount to avoid having to realign optics.The placement of MM2 and MM3 should not affect the incident pump or probe beams in the perpendicular setup. - With a low-profile stirrer (Table of Materials), stir the sample moderately and perform TA measurements. Repeat steps 4.6-4.14.
NOTE: For 1-20 Hz lasers, a lower power can often be used (~1 mJ/pulse).
6. Ultrafast transient absorption measurements (ufTA)
- Aligning pump and probe beams for maximum overlap
- The MOF suspension procedure in section 1 does not change. The pre-TA absorption measurements (section 2) do not change, except using SC2 instead of SC1 (Table of Materials). If needed, the purging process does not change either.
- To align the pump and probe beams for ufTA measurements, begin by preparing a solution of a well-known chromophore [e.g., Ru(bpy)32+] in a 2 mm path length cuvette with an OD of 0.5-1 at the excitation wavelength. There is no need to purge the sample.
NOTE: Choose a standard sample that exhibits a TA spectrum in the same wavelength region as the MOF sample. Oftentimes, the MOF linker can be used as the standard. - Turn on the ultrafast laser pump source and spectrometer (Figure 3). Open up the optical parametric amplifier software (if present) and set it to the desired excitation wavelength. Open up the ufTA spectrometer software and choose a probe window (UV-visible, visible, or near-infrared [near-IR]).
NOTE: Make sure that the optical delay stage is aligned at short and long time delays. Depending on the system, this is done manually or through the spectrometer software. Most commercial systems have an "Align Delay Stage" option in the software that can be clicked to align it.
NOTE: If possible, turn off the lights or minimize light interference when observing the pump and probe beams. - Place the standard cuvette in the sample holder in line with the probe beam. Adjust the pump source power with an ND filter wheel (Figure 3, ufND) to see the pump beam if necessary. Place a white note card against the cuvette side facing the pump and probe beam.
- Adjust the pump spot on the note card with the turning knobs on the kinematic mount, such that it is at the same vertical height as the probe beam, and horizontally adjust the pump so that it is within 1 mm or 2 mm next to the probe beam. Without the note card, fine-tune the vertical and horizontal positions of the pump beam to obtain the highest TA spectral signal.
- Adjust the focus (Figure 3, TS) of the pump beam, so it is at its smallest spot size when hitting a standard sample cuvette. The focus is at the smallest point when the maximum signal is obtained. Once the highest spectral signal is obtained, the pump and probe beams are optimally aligned.
NOTE: Commercial ufTA systems (Table of Materials) typically have a Live View option that allows the user to set the time zero and see the whole TA spectrum before officially measuring the sample.
- Determining the pump beam spot size and energy density
- With the pump and probe beams aligned, replace the sample cell holder with a mounted pinhole wheel (2,000-25 µm holes; Table of Materials) at the focal point of the laser beam (Supplementary Figure 1, PHW). Make sure that the pinhole wheel is nearly (if not exactly) perpendicular to the path of the laser beam.
- Set up the pinhole wheel such that the laser beam is passing through the 2,000 µm pinhole. Set up a detector attached to a power meter (Supplementary Figure 1, PWR) closely on the other side of the pinhole wheel, such that all of the laser beam is hitting the detector.
- Adjust the pump source power with an ND filter wheel so the detector is measuring sufficient power. Note the average power at that pinhole size.
- Rotate the pinhole wheel to a smaller pinhole size and adjust the vertical and horizontal position of the laser beam to attain the maximum power output at that pinhole. Note the power for the pinhole size. Repeat this step with progressively smaller pinholes until the smallest pinhole is reached.
NOTE: While the pinhole measurements are more of an approximate method, it is sufficient enough for measurements when comparing it to the alternative method of using a CCD camera, which can cost thousands of dollars. - In data analysis software, plot the data to generate half of a pseudo-gaussian curve (it is not going to be perfect because the beam inherently is not completely gaussian). To get a symmetrical curve, take the same data and paste it in ascending order of spot sizes.
- Multiply the data by -1, so the minimum is now the maximum. Plot the data and fit it to a gaussian curve. Divide the maximum value of the fitted curved by e2. The width of the curve at 1/e2 is the approximate spot size diameter.
- Linear power response check
- To ensure that no non-linear effects are present at a desired power level (e.g., multiphoton excitation processes, multiparticle decays), the signal at multiple points in the MOF TA spectrum right after the chirp response needs to be recorded at different powers. Determine five power levels to make up a curve.
- Replace the pinhole wheel with the sample holder and place the standard sample back in the holder. Repeat step 6.1 (the realignment process should be much easier as the pump beam was only minorly adjusted in step 6.2).
- Once the pump and probe beams are aligned, and the MOF sample is stirring in the sample holder, measure and record the average pump power with a power meter attached to a detector in the pump beam path.
- Remove the detector from the beam path, and in the Live View TA mode, record the ΔOD signal of the MOF sample at different points in the TA spectrum right after the chirp response (~2-3 ps). Repeat steps 6.3.3 and 6.3.4 at the other four power levels.
NOTE: Sometimes the signal is quite weak at lower power levels, so if the option is available, increase the averaging time in the "live-view" mode to 5-10 s to get a better signal-to-noise ratio and lower the probe beam signal fluctuations. We commonly set the averaging time to 2-5 s throughout all power measurements and record the OD at a wavelength with each subsequent averaging period a few times to get a standard deviation at each power. - Plot the recorded data points as ΔOD versus incident power in data analysis software. If there is a linear power response, the resulting plot forms a straight line, with the y-intercept at zero. If there is a non-linear power response, as expected, significant deviations from a linear curve are typically observed.
- Determining the energy density hitting the suspension sample
- With the pump beam spot size and incident power hitting the MOF suspension known, the approximate energy density can be determined.
NOTE: For example, an approximate spot diameter of 250 µm provides a radius of ~125 µm. After converting the radius to cm, the surface area of the spot can be calculated: A = πr2 = π(0.0125cm)2 ≈ 0.0005 cm2. Dividing the incident power (e.g., 30 µW) by the laser repetition rate (500 Hz) gives an average energy per pulse of 0.06 µJ. Finally, by dividing the average energy per pulse with the spot surface area, an average energy density per pulse of 120 µJ·cm-2 is obtained. The ideal energy density is one that provides an adequate TA signal while falling in a linear range of pump power; however, if a lower power can be used without sacrificing too much signal, it should be used. A ΔmOD of ~1 at <10 ps is a good compromise between signal and pump power.
- With the pump beam spot size and incident power hitting the MOF suspension known, the approximate energy density can be determined.
- Performing ultrafast TA measurements
- With the MOF sample in the holder, pump and probe beams overlapped, and an ideal excitation power chosen for the sample, perform ufTA measurements.
- Check the Live View window and ensure the time zero is correctly set to the start of the detector chirp.
NOTE: When switching between the standard sample and the MOF sample, the time zero may be slightly shifted, hence the need to check again. - Exit out of the Live View window to the main spectrometer software. Ensure that the MOF suspension provides an optimal TA spectrum throughout the scanned time window by setting parameters for a quick scan and clicking the Start button. Typical quick-scan parameters are a time window of -5 ps to 8,000 ps, one scan, 100 data points, an exponential point map (i.e., the 100 data points recorded in increments that fit an exponential curve), and an integration time of 0.1 s.
- Once the quick-scan ufTA spectrum is finished and looks good overall, change the scan parameters for a higher-quality measurement and click the Start button. Typical parameters are a time window of -5 ps to 8,000 ps, three scans, 200-300 data points, an exponential point map, and an integration time of 2-3 s.
NOTE: It is generally advised that the measurement time should not exceed 1 h to avoid prolonged degradation, especially at higher pump powers. - Once the high-quality ufTA spectrum is finished, take the sample out of the sample holder and measure the absorption spectrum of the sample to ensure little degradation. Further confirm minimal degradation by passing the suspension through a 20 nm syringe filter (Table of Materials) and measure the absorption spectrum again.
7. Preparation of MOFs for emission measurements
- Depending on the excitation wavelength, PNH2 emits fluorescence and is consequently omitted from this procedure to obtain the true emission spectra and kinetics of the MOF suspension. Additionally, the syringe filtration process in steps 1.7 and 1.8 is omitted.
NOTE: These omissions do not noticeably impact emission measurements. - Weigh out 1 mg of MOF and transfer it to a clean vial. Transfer 3-4 mL of DMF to the vial containing MOF. Repeat step 1.3.
- Measure the absorbance spectrum of the MOF suspension and dilute the suspension until an OD of 0.1-0.2 is attained at the excitation wavelength (section 2).
- Perform the aforementioned purging procedure (section 3). The MOF suspension is now ready for fluorescence measurements.
8. MOF emission measurements
- Turn on the fluorimeter and arc lamp (Table of Materials, Supplementary Figure 2). Open the fluorimeter software and select the emission mode. Place the purged MOF suspension into the sample holder and moderately stir.
- With the excitation wavelength established in step 7.3, set the excitation and emission monochromator slits to 5 nm as a starting point and perform a cursory emission scan with an integration time of 0.1 s.
- Once the emission bandwidths have been optimized to deliver a good signal (>10,000 counts), measure the MOF emission spectrum using a 1 s integration time (or longer). Then, measure the excitation spectrum of the MOF at a selected emission wavelength. Ensure that the excitation spectrum looks nearly identical to the MOF absorption spectrum.
- Close the arc lamp slit and switch the instrument mode to TCSPC (time-correlated single photon counting) on the software.
- Select one of the LEDs used for TCSPC with the desired excitation wavelength and attach it to a sample chamber window perpendicular to the detector window. Attach the necessary wires to the LED to integrate it into the fluorimeter.
- Set the instrument to the desired emission wavelength, the bandwidth to 5 nm (adjust if necessary), and the time window to 150 ns as a starting point (it can be shortened depending on the sample lifetime). Apply these settings and start the TCSPC measurements from the software window.
NOTE: A general stopping point for most TCSPC measurements is when the maximum counts have reached a value of 10,000. Additionally, the optimal detector counting rate is 1%-5% of the LED repetition rate in order to follow Poisson statistics. Consult the TCSPC LED manufacturer to obtain the device specifications if not already provided.
- Set the instrument to the desired emission wavelength, the bandwidth to 5 nm (adjust if necessary), and the time window to 150 ns as a starting point (it can be shortened depending on the sample lifetime). Apply these settings and start the TCSPC measurements from the software window.
The electronic absorption spectra of PCN-222(fb) with and without PNH2 and filtering are shown in Figure 4. The MOF without PNH2 was just tip-sonicated and diluted. When comparing the two spectra, the biggest difference is the minimization of baseline scatter, which shows up as a broad upward absorption with decreasing wavelengths and also broadens the electronic transitions quite noticeably. For further comparison, the PCN-222(fb) ligand in solution, tetracarboxyphenylporphyrin (H2TCPP), is provided in Supplementary Figure 3. An indicator of baseline scatter is an upward absorption in the MOF where the ligand in solution does not absorb. In the case of TCPP, it has no absorption at 800 nm, whereas the MOF without PNH2 shows a clear "absorption" in this region. One issue sometimes faced is finding the appropriate amount of MOF needed to attain a filtered suspension of sufficient absorbance. This is usually a process of trial and error, but if the filtered MOF suspension absorbance does not change over a range of MOF amounts, then using a syringe filter with slightly larger pores usually works.
Emission measurements of tip-sonicated PCN-222(fb) without PNH2 and H2TCPP in DMF were performed and are shown in Figure 5. Without the use of PNH2, the excitation and emission spectra of PCN-222(fb) and H2TCPP in DMF align quite well, indicating PNH2 is not necessary for these measurements. In our prior work, we attribute the differences in emission lifetimes (Figure 5C) between PCN-222(fb) (1.5 ns, 3 ns) and H2TCPP (4 ns, 12 ns) to energy transfer quenching processes between protonated and unprotonated H2TCPP linkers in the MOF11. If the PNH2 suspension protocol is employed for emission measurements, the PNH2 will emit in the visible region (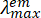 = 475 nm), highlighting its primary setback. Depending on the polymer and concentration, they exhibit absorptions in the UV region and sometimes in the visible region. In the case of PNH2, as shown in Supplementary Figure 4, its absorption onset occurs ca. 450 nm, albeit at a weak level (~0.01 OD). Moreover, when excited by 415 nm light, PNH2 has a broad emission spectrum (Supplementary Figure 5). While PNH2 presents an issue for emission measurements, its involvement with transient absorption measurements is minimal. If a sample needs UV excitation for transient absorption measurements, it is imperative that control experiments with a solution of polymer be performed. In most cases, the polymer TA spectrum (if present) can be subtracted from the MOF spectrum, or their decay lifetimes can be identified within the MOF decay lifetimes. A good rule is keeping the amount of polymer at or below 50 mg per sample.
= 475 nm), highlighting its primary setback. Depending on the polymer and concentration, they exhibit absorptions in the UV region and sometimes in the visible region. In the case of PNH2, as shown in Supplementary Figure 4, its absorption onset occurs ca. 450 nm, albeit at a weak level (~0.01 OD). Moreover, when excited by 415 nm light, PNH2 has a broad emission spectrum (Supplementary Figure 5). While PNH2 presents an issue for emission measurements, its involvement with transient absorption measurements is minimal. If a sample needs UV excitation for transient absorption measurements, it is imperative that control experiments with a solution of polymer be performed. In most cases, the polymer TA spectrum (if present) can be subtracted from the MOF spectrum, or their decay lifetimes can be identified within the MOF decay lifetimes. A good rule is keeping the amount of polymer at or below 50 mg per sample.
Both nsTA and ufTA spectra were obtained with MOF suspensions. In Figure 6 are the TA spectra of PCN-222(fb) with and without PNH2, and H2TCPP in solution right after laser excitation at 415 nm (Soret band excitation). As observed in the spectrum of PCN-222(fb) without PNH2, there is a substantial amount of scatter present, causing the TA spectrum to become increasingly negative with decreasing wavelength. The non-PNH2 TA spectrum (Figure 6A) is in stark contrast to the spectrum of H2TCPP in solution and is a cause for concern. Moreover, the kinetics of H2TCPP and PCN-222(fb) without PNH2 are starkly different (Figure 7). Looking at the spectrum of PCN-222(fb) with PNH2, both the lifetimes and spectra align much better with the H2TCPP TA spectrum11. To gain a complete photophysical picture, a quality initial TA spectrum of the MOF needs to be obtained, along with kinetics at the ground-state bleach (negative signal) and excited state absorptions (positive signal) to see if they agree with one another. Additional measurements using the narrow-angle nsTA setup are presented in Supplementary Figure 6. Comparing the nsTA spectra of PCN-222(fb) between both experimental setups shows a moderate improvement in signal at lower power densities with the narrow-angle setup. Looking at the ufTA spectrum of PCN-222(fb) with PNH2, there is a close resemblance to the linker in solution (Figure 8), showing a ground-state bleach at ~420 nm and excited-state absorptions on either side of the bleach. With both the nsTA and ufTA measurements of PCN-222(fb) with PNH2 in good agreement with H2TCPP in solution, we therefore conclude the observed signal is from the MOF and not due to scatter. After measurements, the absorption spectrum of PCN-222(fb)+PNH2 was remeasured (Supplementary Figure 7) and looked nearly identical to the initial spectrum, indicating minimal degradation throughout the experiment. For further confirmation of any degradation, the MOF suspension can be passed through a 20 nm syringe filter (Table of Materials), and the consequent UV-Vis spectrum of the filtrate should have minimal absorbances from the MOF linker, which would otherwise indicate degradation.
Control experiments and literature on the ligand in solution are key factors when analyzing MOF TA spectra. The broad negative signal observed in MOF TA spectra should be taken as a universal sign that there is excessive scatter occurring from the MOF. Additionally, when looking at the kinetic profile of MOFs with excess scatter arising from both the pump and probe beams, the scatter does not just decay within the instrument response function (IRF; usually the pulse width of the laser); it can have lifetimes up to microseconds that mask the true kinetic decay, however the reason behind this behavior is largely unexplored in the MOF community (Figure 7A). The main takeaway is that, if the signal is broadly negative and the lifetimes are not like those of the ligand (there are exceptions), then the data is not worth interpretation.
Figure 1: Simplified schematic of a perpendicular pump-probe nsTA setup (Table of Materials). P1-P3 are the quartz directional/alignment prisms; CCM1,2 are directional concave mirrors to guide the probe beam; SC1 is the 1 cm sample cuvette used in nsTA measurements; SM1 is the sample mount provided by the spectrometer manufacturer; BD is a beam dump (optional); FL is a focusing lens provided by the instrument manufacturer. To align the pump laser (Actinic Pump) with the probe beam in the sample chamber, the intracavity prism (P3) must be adjusted. All other optics are stationary. Please click here to view a larger version of this figure.
Figure 2: Simplified schematic of a narrow-angle pump-probe nsTA setup (Table of Materials). P1-P3 are the quartz directional/alignment prisms; CCM1,2 are directional concave mirrors to guide the probe beam; SC1 is the 1 cm sample cuvette used in nsTA measurements; SM1 is the sample mount provided by the spectrometer manufacturer; BD is a beam dump (optional); FL is a focusing lens provided by the instrument manufacturer; CCL is a biconcave lens; CVL is a plano-convex lens; MM1-3 are directional mini mirrors to guide the pump beam to the sample cell; SC2 is a 2 mm path length sample cell; SM2 is a clamping sample mount used in ufTA measurements as well. The key factors needed to align the pump and probe beams are proper placement of the pump beam on mirrors MM1-3 and SC2, while SC2 remains at the focal point of the probe beam. Please click here to view a larger version of this figure.
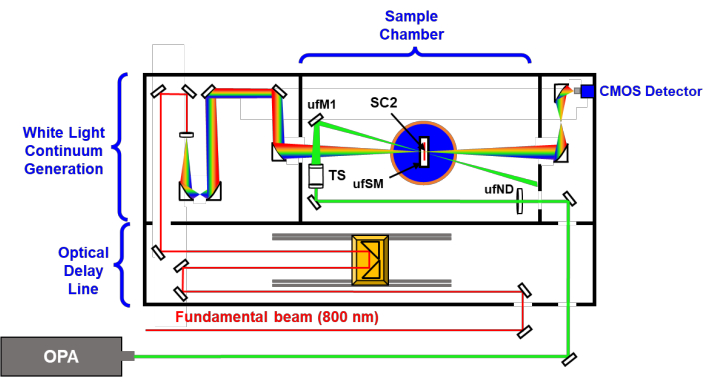
Figure 3: Simplified schematic of the ultrafast transient absorption setup (Table of Materials) used to characterize MOFs. OPA is the optical parametric amplifier used to generate the pump source; ufND is the ND filter wheel used to attenuate the incoming pump power; TS is the telescope used to focus the pump beam; ufM is the kinematic mirror that directs the incoming pump beam onto the sample cell and aligns the pump beam with the probe beam; SC2 is the 2 mm path length sample cell for ufTA measurements; ufSM is a clamping sample mount used in ufTA measurements. The key to aligning pump and probe beams for MOF measurements is first aligning the beams with a dissolved standard sample. Please click here to view a larger version of this figure.
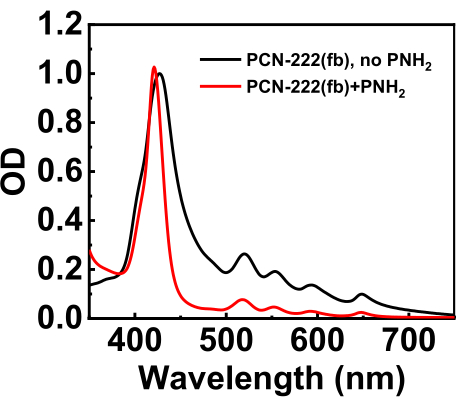
Figure 4: Steady-state absorption spectra of tip-sonicated PCN-222(fb) without PNH2 (black trace), with PNH2 and filtration (red trace), and the absorption spectrum of H2TCPP (MOF linker) shown as the blue trace. The solvent was DMF. A key indicator of scatter is a broad upward absorption underneath the true sample absorption spectrum, as shown in the absorption spectrum of PCN-222(fb) without PNH2. Conversely, the sample with PNH2 barely exhibits an upward absorption. Please click here to view a larger version of this figure.
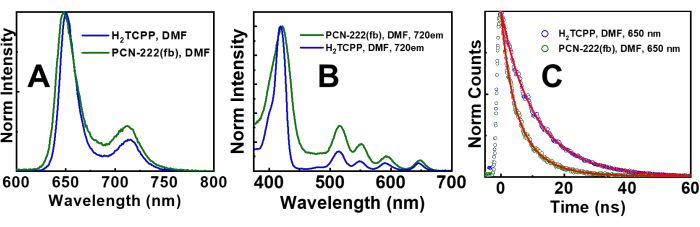
Figure 5: Emission spectra. (A) Emission spectra of tip-sonicated and diluted PCN-222(fb) (green trace) and H2TCPP (MOF ligand; blue trace); (B) Excitation spectra of tip-sonicated and diluted PCN-222(fb) (green trace) and H2TCPP (MOF ligand; blue trace) measured at 720 nm; (C) Time-correlated single photon counting (TCSPC) decay traces of PCN-222(fb) (green trace) and H2TCPP (blue trace) measured at 650 nm. The kinetic fits are the red traces. The solvent was DMF and the excitation wavelength for both spectral and TCSPC emission measurements was 415 nm. The emission and excitation spectra of PCN-222(fb) and H2TCPP align closely with one another, and the kinetic profiles of H2TCPP and PCN-222(fb) are comparable as well. Prior work attributed the shortening of the lifetimes in PCN-222(fb) (1.5 ns, 3 ns) compared to H2TCPP (4 ns, 12 ns) to energy transfer quenching from unprotonated MOF linkers (long lifetime component) to protonated linkers (short lifetime component) that act as energy traps11. This figure has been adapted with permission from Benseghir et al.11. Please click here to view a larger version of this figure.
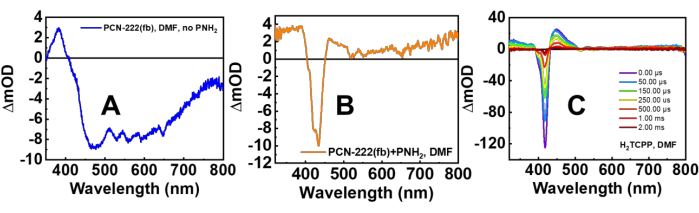
Figure 6: Nanosecond TA spectra. The spectra of tip-sonicated PCN-222(fb) (A) without PNH2, (B) with PNH2 and filtration, and (C) H2TCPP (MOF ligand) in DMF. λex = 415 nm, 3 mJ·cm-2. Similar to the ground-state absorption spectrum of PCN-222(fb) without PNH2, the TA spectrum also shows a broad "absorbance" feature from 450-800 nm attributed to scatter. Comparatively, the TA spectrum of PNH2@PCN-222(fb) resembles that of its parent linker H2TCPP, indicating a genuine TA signal from the MOF. This figure has been adapted with permission from Benseghir et al.11. Please click here to view a larger version of this figure.
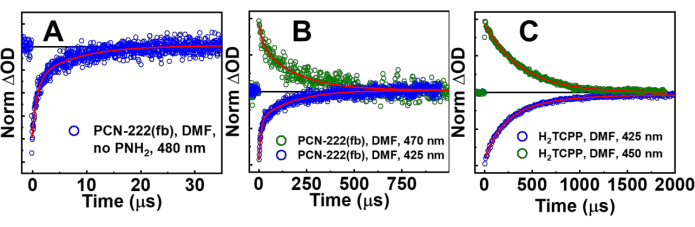
Figure 7: nsTA kinetic decay traces and their fits (red traces). (A) Tip-sonicated PCN-222(fb) without PNH2 at the ground-state bleach (GSB; 420 nm) and excited state absorption (ESA; 385 nm), (B) tip-sonicated and filtered PCN-222(fb) with PNH2 at 419 nm and 470 nm, and (C) H2TCPP (MOF ligand) at 420 nm and 470 nm in DMF. λex = 415 nm, 3 mJ·cm-2. Compared to PCN-222(fb), the kinetic decays of PNH2@PCN-222(fb) align with the time profile of H2TCPP much better. We attribute the decay kinetics observed in PCN-222(fb) to scatter from both the probe and pump beams. It is important to note that scatter can often produce kinetics not just limited to the instrument response time, but additional decays extending into the microsecond region. This figure has been adapted with permission from Benseghir et al.11. Please click here to view a larger version of this figure.
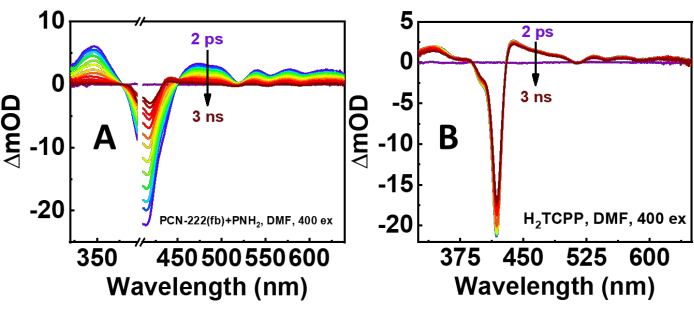
Figure 8: ufTA spectral time mappings (2 ps-3 ns; purple to crimson). (A) Tip-sonicated PCN-222(fb) with PNH2 and (B) the MOF linker H2TCPP in DMF. λex = 400 nm, 50 µJ·cm-2. All the ufTA spectra bear similar features, indicating a genuine signal produced by the MOF. In the case of PCN-222(fb), the spectral changes are more pronounced than the linker alone, which are likely attributed to the quenching of the excited singlet state by efficient energy transfer to protonated H4TCPP centers in the MOF, as well as some energy transfer to the PNH2 suspending agent. The protonated MOF linkers arise from the acidic synthetic conditions needed to make the MOF. Please click here to view a larger version of this figure.
Supplementary Figure 1: Schematic of the ufTA sample chamber when determining the pump laser spot size. ufND is the ND filter wheel used to attenuate the incoming pump power; TS is the telescope used to focus the pump beam; ufM is the kinematic mirror that directs the incoming pump beam onto the sample cell and aligns the pump beam with the probe beam; PHW is the circular pinhole wheel with various hole diameters (Table of Materials); PWR is the power meter used to measure the power at decreasing pinhole sizes. We stress that the pinhole wheel needs to be at the focal point of the pump beam to get accurate spot sizes. Please click here to download this File.
Supplementary Figure 2: Schematic of the fluorimeter used for MOF emission measurements. SC1 is a 1 cm path length sample cell (Table of Materials); FO1 are the excitation wavelength focusing optics; FO2 are the TCSPC (time-correlated single photon counting) LED focusing optics; PMT is a photomultiplier tube for spectral emission measurements. Please click here to download this File.
Supplementary Figure 3: Absorption spectrum of H2TCPP in DMF. The strong absorption at 420 nm is an S0→S2 transition (Soret band), and four vibronic transitions from 500-700 nm are S0→S1 transitions (Q-bands). Please click here to download this File.
Supplementary Figure 4: Absorbance spectrum of PNH2 in DMF. Absorbance onset occurs at ~450 nm. Please click here to download this File.
Supplementary Figure 5: Emission spectrum of PNH2 in DMF when excited by 415 nm light. Because PNH2 fluoresces, we often refrain from using it during emission measurements. Please click here to download this File.
Supplementary Figure 6: Nanosecond TA spectra of tip-sonicated and filtered PCN-222(fb) using a narrow-angle pump-probe setup (see Figure 2 for the schematics). Compared to the conventional perpendicular pump-probe setup, the narrow-angle setup shows a noticeable increase in signal and signal-to-noise ratio using lower pump energies (1 mJ·cm-2). λex = 415 nm. Please click here to download this File.
Supplementary Figure 7: Absorption spectrum of PCN-222(fb)+PNH2. The absorption spectrum before nsTA measurements (red trace), after nsTA measurements (blue trace), and the 20 nm MOF filtrate after nsTA measurements (green trace), indicating little sample degradation over the course of the experiment. This figure has been adapted with permission from Benseghir et al.11. Please click here to download this File.
